Refine search
Actions for selected content:
106129 results in Materials Science
Charles S. Barrett: X-Ray Metallurgist — An Informal Portrait
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 3 / September 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. 163-175
-
- Article
- Export citation
Chemical accuracy and precision in Rietveld analysis: The crystal structure of cobalt(II) acetate tetrahydrate
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 1 / March 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 27-39
-
- Article
- Export citation
X-Ray Powder Structural Analysis of the Complexes of Benzo-15-Crown-5 with NaClO4 and KI: [C14H20O5]NaClO4 and [C14H20O5]2KI
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 2 / June 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. 99-101
-
- Article
- Export citation
Cumulative Author Index
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 1 / March 1997
- Published online by Cambridge University Press:
- 10 January 2013, p. 67
-
- Article
- Export citation
Standardless Quantitative X-Ray Phase Analysis – Estimation of Precision
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 4 / December 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 196-200
-
- Article
- Export citation
International X-ray Analysis Society (IXAS) forms in Barcelona
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 3 / September 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 161-162
-
- Article
-
- You have access
- Export citation
PDJ volume 11 issue 1 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 1 / March 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f12
-
- Article
-
- You have access
- Export citation
Computer Comments
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 3 / September 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 261-263
-
- Article
- Export citation
X-ray powder diffraction data and structural study of Fe2GeSe4
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 4 / December 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. 196-201
-
- Article
- Export citation
Cumulative Author Index
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 3 / September 2000
- Published online by Cambridge University Press:
- 10 January 2013, p. 209
-
- Article
- Export citation
Crystal chemistry of (RE, A)2M3O7 compounds (RE=Y, lanthanide; A=Ba, Sr, Ca; M=Al, Ga)
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 3 / September 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. 195-202
-
- Article
- Export citation
X-ray diffraction data for two new calcium uranium (VI) hydrates
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 2 / June 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 81-86
-
- Article
- Export citation
PDJ volume 8 issue 4 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 4 / December 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f8
-
- Article
-
- You have access
- Export citation
Standard X-Ray Diffraction Powder Patterns of Fifteen Ceramic Phases
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 2 / June 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. 106-117
-
- Article
- Export citation
QPDA – A User-Friendly, Interactive Program for Quantitative Phase and Crystal Size/Strain Analysis of Powder Diffraction Data
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 1 / March 1991
- Published online by Cambridge University Press:
- 10 January 2013, p. 52
-
- Article
-
- You have access
- Export citation
-
Recent developments in the Rietveld method for the analysis of powder diffraction data have seen the method evolve from its original purpose of crystal structure refinement to nclude the determination of phase abundance in polycrysalline mixtures and the estimation of crystal size and strain parameters. However, the Rietveld method is not easy to use and may deter many powder diffractionists, who are not inerested in structure refinement per se, from using the method in its non-structural applications.
In order to overcome the difficulties in using the Rietveld method, a program, QPDA (for Quantitative Powder Diffraction Analysis), has been written that sets the conditions necessary for a single or multi-phase refinement, runs the Rietveld program and extracts phase abundance and size/strain information from the refined parameters. The program comprises a user-friendly, default-driven system of subroutines, written initially in VAX Fortran, and operates from a database of inorganic materials frequently encountered in a wide range of minerals and materials science industries.
Equations (3) and (5) should be corrected to read as follows:
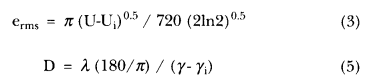
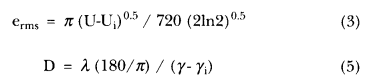
JCPDS-ICDD Subcommittee Activities
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 3 / September 1986
- Published online by Cambridge University Press:
- 10 January 2013, p. 279
-
- Article
- Export citation
X-Ray Peak Intensities for the Binary Compound Al3Ti
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 3 / September 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 165-166
-
- Article
- Export citation
PDJ volume 8 issue 1 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 1 / March 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f10
-
- Article
-
- You have access
- Export citation
X-ray diffraction analysis of the cause of catalyst deactivation
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 1 / March 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 14-17
-
- Article
- Export citation
Powder Diffraction Data for Synthetic Potassium-Richterite, Nickel-Potassium-Richterite and Cobalt-Potassium-Richterite
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 1 / March 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 52-55
-
- Article
- Export citation
