Refine search
Actions for selected content:
106129 results in Materials Science
Calendar of Meetings
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 4 / December 1986
- Published online by Cambridge University Press:
- 10 January 2013, pp. 346-347
-
- Article
-
- You have access
- Export citation
A Simplified Sample Preparation Method for Multiple Guinier Cameras
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 1 / March 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 26-27
-
- Article
- Export citation
Synthesis and X-Ray Powder Diffraction Analysis of a New Metallic (but not Superconducting) Copper Oxide: La1.67Sr0.33Cu2O5
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 81-83
-
- Article
- Export citation
Powder diffraction data and Rietveld refinement of metastable t-ZrO2 at low temperature
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 2 / June 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 96-98
-
- Article
- Export citation
X-Ray Diffraction Analysis of Strontium in Barites
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 2 / June 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 70-73
-
- Article
- Export citation
Structures and X-ray diffraction patterns of compounds in the Sr–Nd–Cu–O system
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 1 / March 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 56-66
-
- Article
- Export citation
Interlaboratory Tests of X-Ray Quantitative Phase Analysis
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 3 / September 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 137-141
-
- Article
- Export citation
Rietveld analysis of high-density polyethylene
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 3 / September 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 217-220
-
- Article
- Export citation
Wavelet denoising of powder diffraction patterns
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 4 / December 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. 300-304
-
- Article
- Export citation
X-ray powder diffraction data for calcium galactarate tetrahydrate CaC6H8O8·4H2O and Rietveld refinement of the crystal structure
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 3 / September 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 175-179
-
- Article
- Export citation
Computer Comments
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 4 / December 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 254-255
-
- Article
- Export citation
Crystal Chemistry and Phase Equilibria Studies of the BaO-R2O3-CuO Systems. II: X-Ray Characterization and Standard Patterns of BaR2O4, R = Lanthanides
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 4 / December 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 187-189
-
- Article
- Export citation
Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 2 / June 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 153-154
-
- Article
-
- You have access
- Export citation
Towards Improved Alignment of Powder Diffractometers
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 1 / March 1986
- Published online by Cambridge University Press:
- 10 January 2013, pp. 26-33
-
- Article
- Export citation
Commercial Announcements
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 1 / March 1993
- Published online by Cambridge University Press:
- 10 January 2013, p. 70
-
- Article
-
- You have access
- Export citation
Evaluation of the detectability and quantification of respirable crystalline silica by X-ray powder diffraction methods
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 4 / December 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 200-227
-
- Article
- Export citation
Powder Diffraction Data of Four Complex Cesium Thiocyanates: Cs3A[B2(SCN)7], with A = Sr, Ba and B = Ag, Cu
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 2 / June 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 111-113
-
- Article
- Export citation
-
Single crystals of Cs3A[B2(SCN)7] with A = Sr, Ba and B = Ag, Cu have been synthesized from aqueous solutions by the evaporation method. The complex thiocyanates are isostructural and crystallize in the tetragonal system with space group
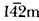 .
.Complete crystal data and optical data for the four compounds are reported. An X-ray powder diffraction pattern for Cs3Sr[Cu2(SCN)7] is given.
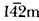 .
.Toward development of an ideal X-ray diffractometer sample holder
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 3 / September 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 200-203
-
- Article
- Export citation
Author Prepared Manuscripts in Computer-Readable form for Powder Diffraction
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 4 / December 1995
- Published online by Cambridge University Press:
- 10 January 2013, p. 235
-
- Article
-
- You have access
- Export citation
New X-ray powder diffraction data for thallium pentaborates Tl[B5O6(OH)4]·2H2O and TlB5O8
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 3 / September 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. 237-239
-
- Article
- Export citation
