Refine search
Actions for selected content:
106129 results in Materials Science
A Tubular Aerosol Suspension Chamber for the Preparation of Powder Samples for X-Ray Diffraction Analysis
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 3 / September 1986
- Published online by Cambridge University Press:
- 10 January 2013, pp. 240-243
-
- Article
- Export citation
Rietveld Structure Refinement of Metastable Lithium Disilicate Using Synchrotron X-Ray Powder Diffraction Data From the Daresbury SRS 8.3 Diffractometer
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 3 / September 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 137-143
-
- Article
- Export citation
Powder X-ray diffraction data for innelite
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 1 / March 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 62-64
-
- Article
- Export citation
Regional Reports
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 1 / March 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 56-59
-
- Article
- Export citation
Subject Classification Scheme Used in Volume 11
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 4 / December 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 324-325
-
- Article
- Export citation
Intensity Calibration Curves for Bragg-Brentano X-Ray Diffractometers
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 2 / June 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 89-94
-
- Article
- Export citation
PDJ volume 13 issue 4 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 4 / December 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f18
-
- Article
-
- You have access
- Export citation
Application of the Rietveld Refinement Procedure in Assaying Powdered Mixtures
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 1 / March 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-6
-
- Article
- Export citation
Crystallographic data about hydrated and anhydrous lithium monoborates
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 3 / September 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 155-159
-
- Article
- Export citation
Crystal Data for CuInTe2
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 1 / March 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 43-46
-
- Article
- Export citation
-
The Debye-Scherrer technique and filtered Cu Kα, radiation were used to obtain powder data for reflections to an angle of 2θ = 155°; a vertical scanning diffractometer was used to obtain intensity data to an angle of 2θ = 99°. The space group is
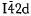 with Z = 4. The lattice parameters of the tetragonal unit cell were measured to be a = 6.189 (1) Å and c = 12.391 (1) Å with c/a = 2.002 The unit cell volume was calculated as U = 474.6 (2) Å3 and the X-ray density as Dx = 6.07 ± 0.003 gm cm−3. By comparing measured and theoretical intensity values, the sublattice distortion parameter x was estimated to be x = 0.227 (12).
with Z = 4. The lattice parameters of the tetragonal unit cell were measured to be a = 6.189 (1) Å and c = 12.391 (1) Å with c/a = 2.002 The unit cell volume was calculated as U = 474.6 (2) Å3 and the X-ray density as Dx = 6.07 ± 0.003 gm cm−3. By comparing measured and theoretical intensity values, the sublattice distortion parameter x was estimated to be x = 0.227 (12).
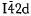 with Z = 4. The lattice parameters of the tetragonal unit cell were measured to be a = 6.189 (1) Å and c = 12.391 (1) Å with c/a = 2.002 The unit cell volume was calculated as U = 474.6 (2) Å
with Z = 4. The lattice parameters of the tetragonal unit cell were measured to be a = 6.189 (1) Å and c = 12.391 (1) Å with c/a = 2.002 The unit cell volume was calculated as U = 474.6 (2) ÅPitfalls in the powder diffraction analysis of zeolites ZSM-5 and ZSM-8
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 3 / September 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 204-212
-
- Article
- Export citation
Residual stresses in a SiC whisker-reinforced alumina composite by high-temperature X-ray diffraction
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 1 / March 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 50-53
-
- Article
- Export citation
Sigmund Weissmann Pioneering X-ray Crystallographer: From insights into the “pathology of structure” (lattice defects) and physical metallurgy came one of the first laboratories for Materials Science—and a life-long association with the PDF
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 4 / December 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 230-238
-
- Article
- Export citation
New Books - Phase Diagrams for Ceramists, Volume VI. Edited by Robert S. Roth, J. R. Dennis and Howard F. McMurdie Contains 696 diagrams of metal oxide systems. 550 pages. Hardbound. - Phase Diagrams for Ceramists, Volume VII. Edited by Lawrence P. Cook and Howard F. McMurdie Contains 933 diagrams of salt systems such as halides, nitrates, sulfates, carbonates, etc. 591 pages. Hardbound.
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 3 / September 1990
- Published online by Cambridge University Press:
- 10 January 2013, p. 174
-
- Article
- Export citation
X-Ray Powder Diffraction Data for β-Lead Telluride
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 4 / December 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. 230-231
-
- Article
- Export citation
-
50-50 atomic percent lead telluride (Altaite), grown by the vapor transport method, was examined with a well aligned Rigaku horizontal beam diffractometer. PbTe is cubic (precise lattice parameter ao = 6.4591(5)Å) with an
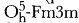 space group and a calculated density of 8.253 g/cm3. Fully indexed powder diffraction data are presented.
space group and a calculated density of 8.253 g/cm3. Fully indexed powder diffraction data are presented.
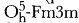 space group and a calculated density of 8.253 g/cm
space group and a calculated density of 8.253 g/cmPDJ volume 7 issue 1 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 1 / March 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b5
-
- Article
-
- You have access
- Export citation
X-Ray Powder Diffraction Analysis of Barium Titanyl Oxalate Tetrahydrate
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 3 / September 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 162-164
-
- Article
- Export citation
Acquisition and Analysis of Powder X-Ray Diffraction Data Using CALS Chromatographic Software
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 3 / September 1986
- Published online by Cambridge University Press:
- 10 January 2013, pp. 222-225
-
- Article
- Export citation
PDJ volume 7 issue 4 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 4 / December 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b3
-
- Article
-
- You have access
- Export citation
Improved powder X-ray data for cancrinites I: Afghanite
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 1 / March 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 68-73
-
- Article
- Export citation
