Refine search
Actions for selected content:
106129 results in Materials Science
Searching and Matching of X-Ray Powder Diffraction Patterns Using a Programmable Calculator
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 3 / September 1986
- Published online by Cambridge University Press:
- 10 January 2013, pp. 235-239
-
- Article
- Export citation
Sodium Copper Oxalate Dihydrate: Na2Cu (C2O4)2. 2H2O Synthesis, Characterization, Morphology and Optical Properties
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 1 / March 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 14-18
-
- Article
- Export citation
X-Ray Powder Diffraction Data for Sodium Octyl Sulfate, Sodium Decyl Sulfate and Sodium Dodecyl Sulfate
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 4 / December 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 244-245
-
- Article
- Export citation
Erratum: “Powder X-ray characterization of natrolite, Na2Al2Si3O10.2H2O’’ [Powder Diffr. 10, 243–247 (1995)]
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 1 / March 1997
- Published online by Cambridge University Press:
- 10 January 2013, p. 53
-
- Article
-
- You have access
- Export citation
Crystal data of Nitrofurantoin C8H6N4O5
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 2 / June 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 107-108
-
- Article
- Export citation
Refinement of unit-cell parameters by whole-powder-pattern fitting technique
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 4 / December 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 272-279
-
- Article
- Export citation
Small-angle scattering of X-rays in a conventional Bragg–Brentano diffractometer for quantitative analysis
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 3 / September 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 170-172
-
- Article
- Export citation
JCPDS-ICDD Subcommittee Activities
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 1 / March 1986
- Published online by Cambridge University Press:
- 10 January 2013, p. 106
-
- Article
- Export citation
Quantitation of crystallinity in substantially amorphous pharmaceuticals and study of crystallization kinetics by X-ray powder diffractometry
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 1 / March 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-6
-
- Article
- Export citation
Données Cristallographiques Sur Le Trioxalato-Chromate(III) D'Ammonium Trihydrate: (NH4)3 (Cr(C2O4)3)· 3H2O
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 2 / June 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. 104-105
-
- Article
- Export citation
-
The trihydrated ammonium trioxalato-chromate(III) (NH4)3 (Cr(C2O4)3)·3H2O crystallizes in the triclinic system with:
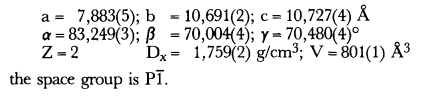 .
.
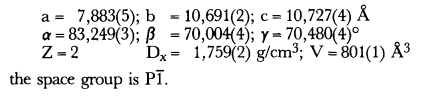
X-ray powder diffraction data for [Co(NH3)5Br]Br2
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 4 / December 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 297-298
-
- Article
- Export citation
Barium hollandite-type compounds BaxFe2xTi8−2xO16 with x=1.143 and 1.333
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 1 / March 1999
- Published online by Cambridge University Press:
- 10 January 2013, pp. 31-35
-
- Article
- Export citation
The quantification of lateritic bauxite minerals using X-ray powder diffraction by the Rietveld method
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 3 / September 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. 136-143
-
- Article
- Export citation
Computer Comments
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 4 / December 1992
- Published online by Cambridge University Press:
- 10 January 2013, p. 243
-
- Article
- Export citation
Crystallographic studies of plant waxes
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 2 / June 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 123-129
-
- Article
- Export citation
Crystal Data for Metal Cimetidine Isotiocionates: M(CM)2(NCS)2 (M = Co(II), Ni(II), Cu(II))
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 4 / December 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 231-235
-
- Article
- Export citation
Correspondent's Reports
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 1 / March 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 54-56
-
- Article
- Export citation
Influence of thermal expansion on the lattice parameter of silicona)
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 4 / December 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 260-264
-
- Article
- Export citation
Incommensurately modulated structure of Bi2Sr2Eu1.3Ce0.7Cu2O10.17 refined from X-ray powder data
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 1 / March 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-6
-
- Article
- Export citation
The automation of DRON diffractometers
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 1 / March 1995
- Published online by Cambridge University Press:
- 10 January 2013, p. 7
-
- Article
- Export citation
