Refine search
Actions for selected content:
106129 results in Materials Science
Design and use of a prototype long-wavelength powder diffractometer
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 2 / June 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 84-88
-
- Article
- Export citation
Book Review - Apparatura i metody rentgenovskogo analiza, Vol. 39 (Instruments and Methods of X-Ray Analysis) Edited by A. I. Slucker, Mashinostroenie, Leningrad, USSR, 1989. 200 pp. (in Russian) - Quantitative Mineral Analysis of ClaysD.R. Peaver and F.A. Mumpton, Editors, The Clay Minerals Society, P.O.Box 880, Evergreen Colorado, 80349 U.S.A. (1990).
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 1 / March 1991
- Published online by Cambridge University Press:
- 10 January 2013, p. 57
-
- Article
- Export citation
Shifts of peak positions due to specimen geometry and beam divergence in X-ray diffractometer
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 4 / December 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 276-280
-
- Article
- Export citation
PACS Headings Used in the Present Index
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 4 / December 1993
- Published online by Cambridge University Press:
- 10 January 2013, p. 259
-
- Article
- Export citation
X-Ray Powder Data for Mellite
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 3 / September 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 172-173
-
- Article
- Export citation
Improved powder X-ray data for cancrinites II: Liottite and sacrofanite
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 1 / March 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 13-19
-
- Article
- Export citation
-
Improved powder X-ray diffraction (XRD) data for liottite and sacrofanite, two members of the cancrinite group of minerals, were obtained using a rotating anode diffractometer. The cell parameters of liottite are a = 12.8575(3), c = 16.0905(6) Å, and the space group is
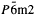 ; the strongest reflections are at 4.834(38), 3.783(12), 3.714(100), 3.312(91), 2.784(13), 2.682(26), 2.470(17), and 2.143(27) Å. The cell parameters of sacrofanite are a = 12.8945(4) and c =74.2128(37) Å, and the possible space groups are P63/mmc, P63mc, and
; the strongest reflections are at 4.834(38), 3.783(12), 3.714(100), 3.312(91), 2.784(13), 2.682(26), 2.470(17), and 2.143(27) Å. The cell parameters of sacrofanite are a = 12.8945(4) and c =74.2128(37) Å, and the possible space groups are P63/mmc, P63mc, and 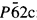 ; the strongest reflections are at 11.18(23), 3.757(25), 3.723(100), 3.488(27), 3.302(62), 2.651(31), 2.645(30), and 2.149(18) Å. The new data include an increased number of indexed peaks and empirical formulae that differ from the reference data (PDF 29-1187 and PDF 35-653, respectively).
; the strongest reflections are at 11.18(23), 3.757(25), 3.723(100), 3.488(27), 3.302(62), 2.651(31), 2.645(30), and 2.149(18) Å. The new data include an increased number of indexed peaks and empirical formulae that differ from the reference data (PDF 29-1187 and PDF 35-653, respectively).
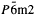 ; the strongest reflections are at 4.834(38), 3.783(12), 3.714(100), 3.312(91), 2.784(13), 2.682(26), 2.470(17), and 2.143(27) Å. The cell parameters of sacrofanite are a = 12.8945(4) and c =74.2128(37) Å, and the possible space groups are P6
; the strongest reflections are at 4.834(38), 3.783(12), 3.714(100), 3.312(91), 2.784(13), 2.682(26), 2.470(17), and 2.143(27) Å. The cell parameters of sacrofanite are a = 12.8945(4) and c =74.2128(37) Å, and the possible space groups are P6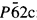 ; the strongest reflections are at 11.18(23), 3.757(25), 3.723(100), 3.488(27), 3.302(62), 2.651(31), 2.645(30), and 2.149(18) Å. The new data include an increased number of indexed peaks and empirical formulae that differ from the reference data (PDF 29-1187 and PDF 35-653, respectively).
; the strongest reflections are at 11.18(23), 3.757(25), 3.723(100), 3.488(27), 3.302(62), 2.651(31), 2.645(30), and 2.149(18) Å. The new data include an increased number of indexed peaks and empirical formulae that differ from the reference data (PDF 29-1187 and PDF 35-653, respectively).Low- and high-temperature structures of neopentylglycol plastic crystal
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 2 / June 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 109-117
-
- Article
- Export citation
-
Plastic crystals, such as neopentylglycol, 2, 2-dimethyl-1,3-propanediol, that exhibit polymorphic behavior are emerging materials for thermal energy storage. The energy is stored isothermally in the γ phase, FCC, during solid-state phase transformations. This γ phase of NPG has been determined as an orientational disordered phase. The low temperature α phase structure, which is of great significance in the evaluation of lattice expansions and other parameters, was first determined in 1961. However, the reported unit cell dimensions and the intensities of the reflections led to erroneous indexing of the powder patterns in binary systems. The α phase structure is redetermined here as monoclinic, M= 104.15 amu, space group P21/n (an alternate setting of
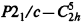 , space group No. 14), a = 5.979(1)Å, b= 10.876(2)Å, c=10.099(2)Å, β=99.78(1)°, V=647.2(2)Å3 at 20°(± 1)C, Dx= 1.069 g cm s−3 for Z=4. In this paper the redetermined structure of the α phase of NPG is presented in projections of the atomic positions, in tables, and in calculated powder pattern and these results are compared with those reported by others. The powder patterns obtained from the Bragg–Brentano diffractometer are compared with our calculated pattern from the single crystal data. The structural parameters of the high temperature phase of NPG as determined by a Guinier diffraction system are also reported.
, space group No. 14), a = 5.979(1)Å, b= 10.876(2)Å, c=10.099(2)Å, β=99.78(1)°, V=647.2(2)Å3 at 20°(± 1)C, Dx= 1.069 g cm s−3 for Z=4. In this paper the redetermined structure of the α phase of NPG is presented in projections of the atomic positions, in tables, and in calculated powder pattern and these results are compared with those reported by others. The powder patterns obtained from the Bragg–Brentano diffractometer are compared with our calculated pattern from the single crystal data. The structural parameters of the high temperature phase of NPG as determined by a Guinier diffraction system are also reported.
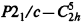 , space group No. 14), a = 5.979(1)Å, b= 10.876(2)Å, c=10.099(2)Å, β=99.78(1)°, V=647.2(2)Å
, space group No. 14), a = 5.979(1)Å, b= 10.876(2)Å, c=10.099(2)Å, β=99.78(1)°, V=647.2(2)ÅCommercial Announcements
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 2 / June 1989
- Published online by Cambridge University Press:
- 10 January 2013, p. 128
-
- Article
-
- You have access
- Export citation
X-ray powder diffraction data for some manganese phosphates and arsenates
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 3 / September 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 155-159
-
- Article
- Export citation
ICDD Crystallographic Scholarship Awards
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 1 / March 1992
- Published online by Cambridge University Press:
- 10 January 2013, p. 60
-
- Article
- Export citation
Crystal Data for [Cu(C15H11N3)X2]·nH2O [X−=NCO (n=1); NCSe and N3 (n=ø)] Compounds
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 3 / September 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 162-164
-
- Article
- Export citation
An Inexpensive Automation of a Powder Diffractometer
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 91-92
-
- Article
- Export citation
X-Ray Powder Diffraction Data for AITh2(VO4)3Compounds with AI= Li, Na, Ag
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 4 / December 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 219-220
-
- Article
- Export citation
Solving the Crystal Structure of Cd5(OH)8(NO3)2·2H2O from Powder Diffraction Data. A Comparison with Single Crystal Data
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 1 / March 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 10-15
-
- Article
- Export citation
Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 3 / September 1986
- Published online by Cambridge University Press:
- 10 January 2013, p. 281
-
- Article
-
- You have access
- Export citation
Studies of Distortion in Potassium Manganese Fluoride, KMnF3, at 10°K: an Example of Powder Profile Analysis Using the Pearson Type VII Distribution
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 1 / March 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 41-43
-
- Article
- Export citation
Subject Index to Volume 9
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 4 / December 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 296-298
-
- Article
- Export citation
PDJ volume 12 issue 2 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 2 / June 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b3
-
- Article
-
- You have access
- Export citation
V. A. Frank-Kamenetsky 1915–1994
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 2 / June 1995
- Published online by Cambridge University Press:
- 10 January 2013, p. 143
-
- Article
-
- You have access
- Export citation
Structural data of the tysonite-type superstructure modification β-PrF3 from X-ray powder and electron single-crystal diffraction
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 1 / March 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 44-46
-
- Article
- Export citation
-
The crystal structure of the tysonite-type superstructure of β-PrF3 has been studied by X-ray powder diffraction and high-energy electron diffraction from single crystals. The crystal data are: a = 7.0795(1) Å, c = 7.2380(2) Å, Z = 6, hexagonal system, space group
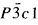 (No. 165), ρcalc = 6.28 g cm−3. Atomic parameters and interatomic distances are presented from the final refinement with Rconv = 1.72%.
(No. 165), ρcalc = 6.28 g cm−3. Atomic parameters and interatomic distances are presented from the final refinement with Rconv = 1.72%.
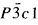 (No. 165), ρ
(No. 165), ρ