Refine search
Actions for selected content:
4288 results in Numerical analysis and computational science
9 - Point-particle methods for disperse flows
-
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 282-319
-
- Chapter
- Export citation
Acknowledgments
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp x-xii
-
- Chapter
- Export citation
7 - Boundary integral methods for Stokes flows
-
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 193-236
-
- Chapter
- Export citation
8 - Averaged equations for multiphase flow
-
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 237-281
-
- Chapter
- Export citation
1 - Introduction: A computational approach to multiphase flow
-
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 1-18
-
- Chapter
- Export citation
References
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 436-465
-
- Chapter
- Export citation
Index
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 466-470
-
- Chapter
- Export citation
3 - Immersed boundary methods for fluid interfaces
-
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 37-77
-
- Chapter
- Export citation
Contents
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp v-vi
-
- Chapter
- Export citation
11 - Coupled methods for multifluid models
-
-
- Book:
- Computational Methods for Multiphase Flow
- Published online:
- 07 December 2009
- Print publication:
- 29 March 2007, pp 386-435
-
- Chapter
- Export citation
First-order semidefinite programming for the two-electron treatment ofmany-electron atoms and molecules
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 249-259
- Print publication:
- March 2007
-
- Article
- Export citation
Sparse grids for the Schrödinger equation
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 215-247
- Print publication:
- March 2007
-
- Article
- Export citation
Diffusion Monte Carlo method: Numerical Analysisin a Simple Case
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 189-213
- Print publication:
- March 2007
-
- Article
- Export citation
Best N-term approximation in electronic structure calculations. II. Jastrow factors
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 261-279
- Print publication:
- March 2007
-
- Article
- Export citation
-
We present a novel application of best N-term approximation theoryin the framework of electronic structure calculations. The paper focusses on thedescription of electron correlations within a Jastrow-type ansatz for thewavefunction. As a starting point we discuss certain natural assumptions onthe asymptotic behaviour of two-particle correlation functions $\mathcal{F}^{(2)}$
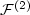 near electron-electron and electron-nuclear cusps. Basedon Nitsche's characterization of best N-term approximation spaces $A_{q}^{\alpha}(H^{1})$
near electron-electron and electron-nuclear cusps. Basedon Nitsche's characterization of best N-term approximation spaces $A_{q}^{\alpha}(H^{1})$ 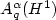 , we prove that $\left.\mathcal{F}^{(2)}\inA_{q}^{\alpha}(H^{1})\right.$
, we prove that $\left.\mathcal{F}^{(2)}\inA_{q}^{\alpha}(H^{1})\right.$ 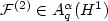 for q>1 and $\alpha=\frac{1}{q}-\frac{1}{2}$
for q>1 and $\alpha=\frac{1}{q}-\frac{1}{2}$ 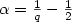 with respect to a certain class of anisotropic wavelet tensor product bases.Computational arguments are given in favour of this specific class compared toother possible tensor product bases. Finally, we compare the approximationproperties of wavelet bases with standard Gaussian-type basis sets frequentlyused in quantum chemistry.
with respect to a certain class of anisotropic wavelet tensor product bases.Computational arguments are given in favour of this specific class compared toother possible tensor product bases. Finally, we compare the approximationproperties of wavelet bases with standard Gaussian-type basis sets frequentlyused in quantum chemistry.
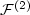
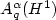
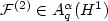
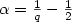
Preface
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 187-188
- Print publication:
- March 2007
-
- Article
- Export citation
Molecular Simulation in the Canonical Ensemble and Beyond
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 333-350
- Print publication:
- March 2007
-
- Article
- Export citation
Atomistic to Continuum limitsfor computational materials science
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 391-426
- Print publication:
- March 2007
-
- Article
- Export citation
Theoretical and numerical comparison of some sampling methods for molecular dynamics
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 351-389
- Print publication:
- March 2007
-
- Article
- Export citation
Converging self-consistent field equations in quantum chemistry – recent achievements and remaining challenges
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 281-296
- Print publication:
- March 2007
-
- Article
- Export citation
Multiscale Materials Modelling: Case Studies at the Atomistic and Electronic Structure Levels
-
- Journal:
- ESAIM: Mathematical Modelling and Numerical Analysis / Volume 41 / Issue 2 / March 2007
- Published online by Cambridge University Press:
- 16 June 2007, pp. 427-445
- Print publication:
- March 2007
-
- Article
- Export citation
