When Lia was about three months old, her older sister Yer slammed the front door of the Lees’ apartment. A few moments later, Lia’s eyes rolled up, her arms jerked over her head, and she fainted. The Lees had little doubt what had happened. Despite the careful installation of Lia’s soul during the hu plig ceremony, the noise of the door had been so profoundly frightening that her soul fled her body and became lost. They recognised the resulting symptoms as quag dab peg, which means ‘the spirit catches you and you fall down’.
The Calm Before the Storm
The above quote comes from a book written about the experience of a family with a child who has epilepsy, and the clash of cultures between their traditional beliefs as an indigenous people from South East Asia and the world of modern (Western) medicine. There are an estimated 60 million people living around the world with epilepsy. In some respects, epilepsy is one of the ‘oldest’ brain diseases. There is a detailed description of epilepsy in one of the collection of Babylonian tablets at the British Museum, which dates to the middle of the first millennium BCE. Translated, it gives clinical observations of what we would recognise as the main seizure types and some of the different epilepsy syndromes. There are written accounts of people with epilepsy, thoughts on the causes and suggestions of treatments that date back centuries. In the past, epilepsy was called the ‘sacred disease’ on account of it being thought to be owing to divine or malign intervention. Hippocrates attempted to dismiss the idea of it being brought on by divine interference, reasoning that it was a disease like any other. There is a long and tragic history of the mistreatment of people with epilepsy; they were often blamed and tortured for being possessed by devils. To this day, it remains one of the most stigmatised diseases and surveys of attitudes towards epilepsy continue to find high levels of misunderstanding of what it is and what causes it. I have spent most of my career investigating the molecular differences found in brain tissue samples from people who underwent neurosurgery to relieve treatment-resistant epilepsy. What my studies over the past decade have shown, along with research by many others, are extensive changes to the microRNA system in the parts of the brain from which seizures arise or to which they spread. We have found changes to the microRNA biogenesis machinery and levels of dozens of microRNAs. This drives adjustments to genes and pathways that are responsible for the control of brain excitability. Some of these represent beneficial adaptations, an effort by cells and networks to keep stable. But other changes are maladaptive, contributing to the problem and driving and sustaining epilepsy, as well as compromising the effectiveness of medicines. This chapter will delve into this brain disease and the discoveries made about microRNAs. The applications of this knowledge include potential future medicines and diagnostic tests.
So, what is epilepsy and what causes it? It is a brain disease whose primary symptom is the recurrence of seizures.[Reference Browne and Holmes205–Reference Devinsky, Vezzani, O’Brien, Jette, Scheffer and de Curtis208] Seizures are brief disturbances of brain function in which groups of neurones or entire brain regions undergo highly synchronous, high-frequency neuronal discharges. The synchrony becomes an electrical storm that temporarily disrupts the function of any brain region in which it arises or which it passes through. What is experienced or observed from the outside depends on where the seizure is occurring. Seizures in a part of the brain that processes sensory information may cause unusual tastes and smells, or visual or auditory disturbances. If a seizure spreads to the motor cortex, the part of the brain that connects to the neurones that contact your skeletal muscles, it will trigger sequential stiffening and contractions of the muscles of the limbs and trunk. If you have ever witnessed a seizure, this is probably the type of seizure that occurred. You may have witnessed other seizures without knowing it. Many types of seizure do not give rise to such physical changes and would be unremarkable to the untrained eye. Petit mal epilepsy, now called absence epilepsy, is an example of this type of epilepsy. Seizures in this form of epilepsy are very brief and cause only temporary disruption of awareness.
We do not fully understand what causes a seizure. At a very simple level, it is because a tipping point is breached between excitation and inhibition in the brain. There is either too much excitatory drive or too little inhibitory drive or a mix of the two. This idea comes from several sources of evidence: from studying brain samples from people who have epilepsy, the genes found to be linked to the disease, electrical recordings and imaging of the brain during a seizure, and our understanding of the mechanisms of action of various drugs that prevent, and neurotoxins that promote, seizures. Let’s start with the last of these. If we give an animal such as a mouse a drug that repeatedly activates glutamate receptors to boost excitatory neurone activity, a seizure will develop within a few minutes. If we block inhibitory neurone activity in the brain with other drugs, a seizure will also develop within a few minutes. Some of the genes in which errors have been found in people with epilepsy encode components of the GABA inhibitory neurotransmission system. Dravet syndrome is an example of a ‘genetic’ epilepsy caused by errors in the DNA sequence that encodes a sodium channel that is required for inhibitory neurones to fire at high frequencies. There are also genetic causes of epilepsy that are owing to errors in genes that work during brain development to coordinate genes switching on and off. Mutations in these can give rise to wiring errors that misplace inhibitory neurones in circuits. Other forms of epilepsy appear to have a less important genetic component or none at all. Epilepsy can arise, for example, owing to injuries to the brain and infections and is more common in old age and in Alzheimer’s disease.[Reference Sen, Jette, Husain and Sander209] In fact, epilepsy can arise at any time of life.
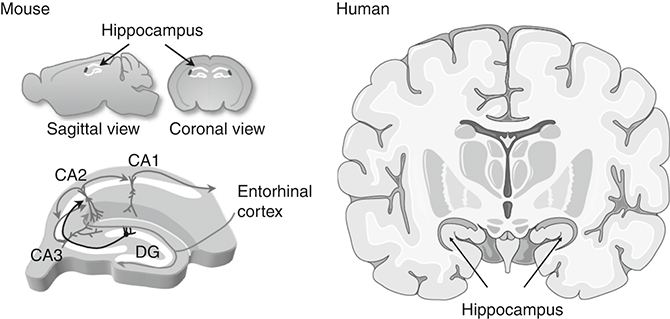
Temporal lobe epilepsy (TLE) is the most common form of treatment-resistant epilepsy in adults. We met this structure in Chapter 5. The temporal lobe is responsible for a number of brain functions including processing sensory information; aspects of spatial navigation, so we know where we are in the world and can remember how to get back home; and the creation, storage and retrieval of certain types of memory, in particular episodic memories such as remembering your first day at school, and emotional memories. Deep within this lobe is the hippocampus. Figure 6.1 shows where this structure lies within the mouse and the human brain and the basic neural circuitry through which information flows.
This shows the location of the hippocampus in the mouse and human brain (in cross-section). The image at the bottom left is a schematic of the circuit connections between the sub-domains called the dentate gyrus (DG), CA1, CA2 and CA3; CA stands for cornu ammonis.
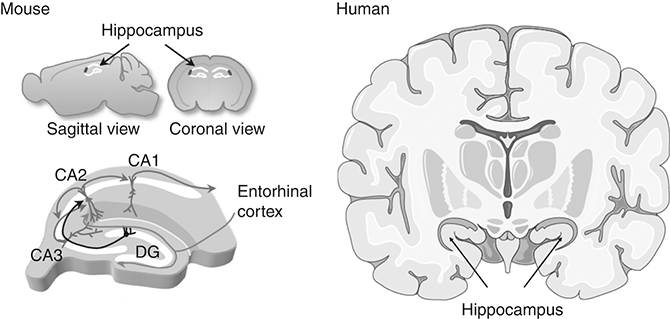
The hippocampus is one of the most studied brain structures. Neuroscientists are very fond of the hippocampus. It has an elegant organisation comprising a three-part circuit through which information flows. Input to the structure comes mainly from a part of the temporal lobe called the entorhinal cortex via a bundle of axons called the perforant pathway. These synapse onto the first part of the hippocampus called the dentate gyrus. This is almost entirely composed of cells called dentate granule neurones. The axons of these neurones form synapses and activate large pyramidal neurones within a subpart of the hippocampus called the CA3 subfield. The CA3 neurones then target another set of pyramidal neurones in a region called the CA1. Finally, the axons of those neurones take signals out of the hippocampus and on to other connected regions of the brain. Interspersed with this circuit are inhibitory neurones that function to integrate signals and regulate firing of the excitatory, or principal, neurones. The hippocampus is also densely packed with all the major types of supporting glia cells.
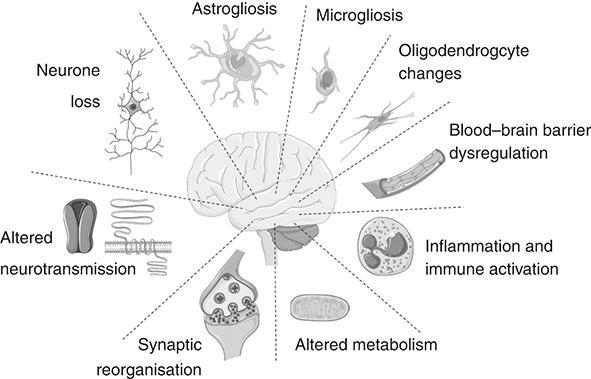
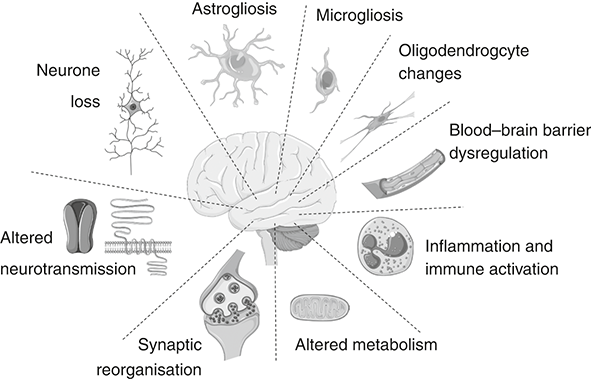
What was noticeable to the first pathologists who studied the brains of people with TLE was that the hippocampus was shrunken and hard.[Reference Sommer211] The shrinking reflected the loss of many neurones and the hardening reflected a scar formed by glial cells. Later, neurosurgeons began removing the hippocampus from patients with treatment-resistant epilepsy and found that this was highly effective at reducing or stopping seizures.[Reference Asadi-Pooya and Rostami212] This proved that the damaged hippocampus was either the source of the seizures or an important part of the circuit. Neurone loss or the accompanying scarring process was suspected to cause this form of epilepsy. Both excitatory and inhibitory neurones have been found to be missing in resection specimens, which may tip the circuit towards seizures either because there are fewer inhibitory neurones or because there is a loss of excitatory neurones that activate the inhibitory neurones. This idea has been extensively supported using animal models of epilepsy. Electrical or chemical stimulation of the hippocampus of adult rats or mice causes neurone loss and, within a few days, epileptic (i.e. spontaneous) seizures. A recent study used an elegant technique to selectively delete just a single subtype of inhibitory neurone that releases a neurotransmitter called substance P in the hippocampus of mice. This caused epilepsy in all the animals within a few days.[Reference Chun, Bumanglag, Burke and Sloviter213] Interestingly, when the brains of the mice were studied months later, many neurones besides the substance P inhibitory neurones were also missing. This proved two fundamental points. An insult to the brain that causes even subtle loss of inhibitory tone in the hippocampus is sufficient to cause TLE. Second, repeated epileptic seizures can cause further damage to the hippocampus that, over time, develops into the same pattern we see in patients with lifelong TLE. The rapid development of epilepsy in some models appears incongruous with most clinical observations, however. Our own studies have found that epilepsy in mice can emerge within two or three days from an inciting event, but many people have a first seizure out of the blue and quite separate in time from any conceivable brain insult. This suggests that the time frame for epileptogenesis (literally, the ‘birth’ of epilepsy) might be different in humans, perhaps because of species differences in cell types, cell density or other factors. Or, an initial insult seeds the epileptogenesis process deep within the brain and the time taken before the first clinical – that is, visible – seizure emerges reflects sub-clinical events that, over time, recruit more brain until a breakthrough seizure happens. The birth of TLE may occur a long time before it shows itself to the outside world. The debate over whether neurone loss is the most important cause of TLE is not settled. Added to which, surgical resection specimens from patients with long-standing TLE are occasionally obtained which lack any obvious neurone loss. In fact, the most consistent finding in the hippocampus of TLE patients is actually extra or expanded glial cells (‘gliosis’).[Reference Blumcke, Spreafico, Haaker, Coras, Kobow and Bien214] And neurone loss and gliosis are not the only pathologies found in resected TLE samples. Several other changes are also notable in most resection specimens. These include neuroinflammation, changes to the dendrites and axons, changes to the vascular supply, including leaky blood vessels, and changes to the extracellular environment, a matrix of proteins and other molecules that create structure for tissue. Experiments in rodents have shown that triggering any one of these processes is sufficient to create a TLE-like syndrome. It is this complex set of pathophysiological processes that makes microRNA-based treatments a good fit for treating TLE (see Figure 6.2).
Pathophysiology of temporal lobe epilepsy
Examples of the various cellular alterations implicated in triggering seizures in TLE.
The Beginning and the End
Despite the enduring changes in the brain, be it pathology or a gene defect, seizures are usually rare and brief events in people with epilepsy. Most of the time, the brain circuits are stable. So, what happens during transitions from ‘no seizure’ to ‘seizure’? When seizures do occur, they are usually brief, so what makes a seizure stop? For some time it was thought that seizures began because inhibitory restraint on the brain network was temporarily lost. Several mechanisms for this onset moment are known and they are not mutually exclusive.[Reference Blauwblomme, Jiruska and Huberfeld215, Reference Glykys, Dzhala, Egawa, Kahle, Delpire and Staley216] There is broad agreement that it involves temporary changes to the inner chemistry of neurones that affect their ability to fire action potentials. For example, inhibitory neurones mainly use the neurotransmitter GABA. When released, GABA binds to an excitatory neurone and opens a gate for the movement of the chloride ion. This ion is more concentrated outside the cell and carries a negative charge. Chloride entry makes the inside of the neurone more negative. Since action potential firing requires a change to a more positive membrane potential, entry of chloride effectively halts the firing of a neurone. But if these gates remain open too long then chloride will build up inside cells. This reduces the natural gradient that favours further chloride entry. At some point we reach a balance and chloride will no longer enter the neurone; it may even reverse and start to leave the neurone. If this happens, we have lost inhibitory tone, and excessive firing and synchronisation of surrounding excitatory (principal) neurones can occur. Another mechanism, described in Chapter 5, is via disruption of the potassium gradient. If potassium accumulates outside the cell, this reduces or depolarises the cell and neurones can lose the ability to reset their membrane potential between firing action potentials. This is a state called depolarisation block and if this happens to an inhibitory neurone then it will impair inhibitory control of the network. Experiments show that resetting inhibitory neurones back to their more negatively charged state allows them to restart firing and restore inhibitory balance. A mixture of these and other mechanisms underlies the neuronal synchronisation that signals the network shifting into a seizure. However, it is incorrect to think that seizures occur because inhibition fails. Human and animal studies in 2018 revealed that, counter-intuitively, GABA-releasing inhibitory neurone firing rates sharply increase immediately prior to seizure onset and may be the trigger for the subsequent synchronisation of excitatory (principal) neurones.[Reference Elahian, Lado, Mankin, Vangala, Misra and Moxon217, Reference Miri, Vinck, Pant and Cardin218]
A seizure, whether it starts in one part of the brain and remains localised, or spreads, or begins on both sides of the brain at once, usually stops after one or two minutes. Why? One of the mechanisms is the build-up of adenosine outside neurones. This is a break-down product of ATP, the ATP having been co-released with many neurotransmitters. Receptors for adenosine are present at the terminals of excitatory neurones; when activated, they prevent further neurotransmitter release. We know that this helps to terminate seizures because blocking the formation of adenosine or the receptors it binds to causes seizures to continue for longer.[Reference Boison219] A second important seizure shut-down mechanism comes from the actions of protons (hydrogen ions/H+). This happens through the build-up of lactic acid and carbon dioxide (which forms a weak acid when dissolved in water) that accompanies metabolic activity during intense neuronal activity. The mechanism of this seizure-terminating effect is via acid-sensing ion channels. The binding of protons to the surface of these receptors causes them to open and allows sodium and calcium ions into the cell. The entry of calcium triggers neurotransmitter release. Because these specialised channels are present on inhibitory neurones, the build-up of acid activates inhibition in the brain. Experiments show that blocking these channels in animals results in seizures that fail to terminate.[Reference Ziemann, Schnizler, Albert, Severson, Howard and Welsh220] Interestingly, acidosis brought on by inhaling carbon dioxide was known to inhibit seizures in people nearly a century ago; if some people with epilepsy hyperventilate and thus blow off too much carbon dioxide, making themselves slightly alkaline, this can bring on a seizure.
Treating Epilepsy
We can prevent seizures in people with epilepsy using drugs called anti-seizure medicines (ASMs). There are more than 30 available. But it was not always so. An account of the treatment of a person with nocturnal epilepsy called Janet Duncan, admitted to Glasgow Royal Infirmary in the 19th century, captures the awful reality of treatment efforts before the modern era of ASMs.[Reference Orr221] It reveals how poorly understood the disease was among health professionals, and how far we have come. Reported in History of a case of epilepsy, Edinburgh Medical and Surgical Journal, vol. 77, pp. 10–23, in January 1852, part of the description is as follows: ‘on the 11th of September she had a number of severe attacks. Dr Alison saw her and bled her; ordered tartar emetic ointment to be rubbed on the head; gave her tartar emetic internally, and also pills, containing extract of colocynth, hyoscyamus, and croton oil.’ Among the multitude of medicines and procedures given to the woman during admission was bleeding by leeches, croton oil to blister the skin on the head (that was tried several times), indigo powder, turpentine, a warm foot bath, wine (at one point a glass was ordered to be taken every hour), enemas (in general a lot of attention to bowel movements – both natural and medically induced) and tartrate of antimony – probably as an emetic (this was both given to her as a drink and rubbed on her head). She later died. If I had to guess, from human (iatrogenic) causes rather than the epilepsy.
The organised and systematic discovery of ASMs began in the twentieth century. One of the earliest drugs for epilepsy and one still in use today is phenobarbital. We have had this for 100 years. It boosts the amount of chloride that GABA causes to flow into neurones. There are other drugs that reduce sodium entry through channels, required during the first phase of an action potential, so effectively they work to temper the frequency of action potentials. Another drug, levetiracetam, modifies the mechanism by which neurotransmitters are released. Some ASMs work through a combination of actions. But there are problems with ASMs. First, not all patients get relief from their seizures. In fact, only about two out of three people with epilepsy achieve seizure control. Disappointingly, and despite the addition of further ASMs over the years, you are statistically as likely to be seizure-free today, in 2023, as you were in 1983.[Reference Chen, Brodie, Liew and Kwan222] Second, ASMs must be taken every day and often several times a day. Regular dosing is needed because our bodies rapidly break down and expel ASMs. Third, ASMs cause serious side effects for many people. Because they mainly work by dampening down brain excitability, patients often experience tiredness and a type of brain fog. There are other side effects, including liver and kidney toxicity. Some ASMs can trigger changes to mental state, including psychosis-like reactions. There are also rare immune reactions. One of these is called Stevens–Johnson syndrome, a skin reaction in certain individuals given the drug carbamazepine that can be life-threatening. Fortunately, a variant in a gene was discovered that predisposes people of Chinese ethnicity and different variants that predispose people of European ancestry. Warnings of these are now included on the medicine box and gene testing can be offered ahead of prescribing.
The other big problem is that ASMs do not cure epilepsy. They merely mask the symptoms – seizures. If you stop taking the medicine the seizures will return, regardless of how many years you have been taking the drug; ASMs do not seem to correct the underlying circuit faults. If we are to cure epilepsy, we need something that actually ‘corrects’ the brain network. This brings us to microRNAs. Could they offer a way to treat or prevent epilepsy? Could blocking microRNAs, and by doing so altering the functions of gene networks, be a way to treat a disease of brain networks? Which epilepsies would this work for?
Why MicroRNAs Make Sense in Temporal Lobe Epilepsy
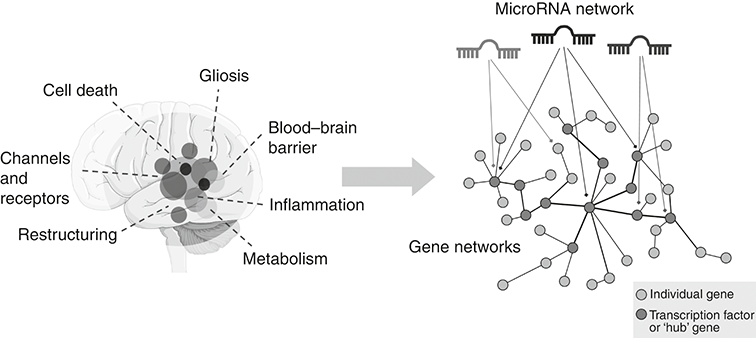
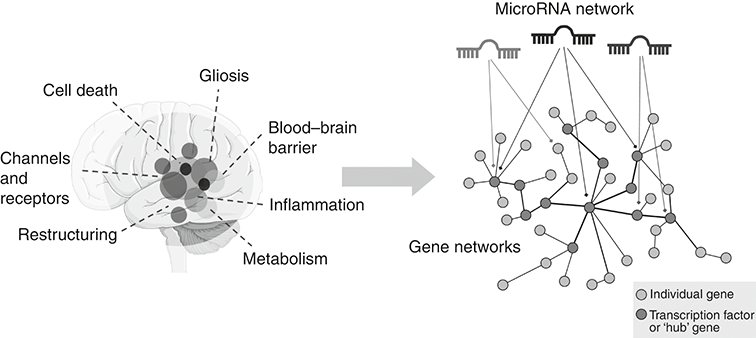
Whether or not the inciting event is neurone loss or something else, most brain tissue from people with treatment-resistant TLE displays complex, multi-factor pathology. Replacing neurones is not easy, although researchers are exploring this, and while neurone loss might have been the sole inciting factor, the brain has usually changed extensively since and it may require changes to many processes to bring it back towards a stable network state. And layered on top of the enduring hyperexcitability is the problem of drug-resistance. Research shows that this is owing to a combination of changes in the movement of substances between the blood and the brain, as well as transporters and glial function, although the exact cause remains unknown. Disease modification will require the ability to adjust several processes and this is where microRNA come in. If a microRNA controls a series of pathways that are causing hyperexcitability and the pathophysiological processes that become altered in TLE, then there is a chance that targeting those – restoring levels if they are deficient or blocking them if they are in excess – could be a new therapeutic approach. Figure 6.3 outlines this idea of microRNAs as being the right fit for a network disorder such as TLE. This concept can be exploited therapeutically, correcting multiple pathways by targeting the microRNAs that shape them; the approach is termed ‘network therapeutics’.
MicroRNAs control gene networks in temporal lobe epilepsy
The schematic on the left shows the temporal lobe and a selection of seizure-generating processes. These are in turn underpinned by altered gene expression networks (right). MicroRNAs regulate multiple targets within these gene networks, acting alone or in combinations on individual transcripts or more strongly via targeting gene control nodes. Darker lines between genes indicate stronger regulatory effects.
Epilepsy and MicroRNAs
My laboratory started working on microRNAs in 2008. This was initiated by a brilliant neuroscientist with a flair for molecular biology, Eva Jimenez-Mateos. We were assisted by a team in the adjacent lab who had just discovered a link between microRNAs and the childhood cancer called neuroblastoma.[Reference Welch, Chen and Stallings223] They generously shared some of their ‘kit’ and know-how, including a way to screen for microRNAs in tissue samples. Unknown to us at the time, a couple of other labs had also had the same idea. Three papers appeared within the space of a few months at the end of 2009 and in early 2010. The first of these, by a team at the University of California at Davis, found changes to microRNA levels in brain and blood samples from rats that were subject to different types of brain injury.[Reference Liu, Tian, Ander, Xu, Stamova and Zhan224] One of the insults was a prolonged seizure, triggered by an injection of a chemical called kainic acid. This is one of the most potent neurotoxins known. It was isolated from a red alga by a Japanese team in 1953. Tiny amounts are sufficient to kill neurones. The reason it is so toxic is that it activates one of the major types of receptor for excitatory neurotransmission in the brain. The receptor, known for a long time as the ‘KA receptor’ but now appropriately assigned another gene name, normally opens in response to glutamate. This causes a pulse of sodium ions to enter the neurone. Kainic acid mimics this, but, unlike glutamate, it is not broken down or removed quickly as a natural neurotransmitter would be, and it lingers, causing repeated activation of the neurone. Sodium overload begins and this then triggers osmotic swelling (water drawn in by salt); cascading secondary processes further damage the cell and within hours the cell is irreversibly damaged or dead. In addition to this direct toxicity, the kainic acid triggers waves of excitation that pass through the brain. This can create a permanently hyperexcitable state. The Davis team took samples from a group of rats that had been injected with kainic acid and measured the levels of microRNAs in the hippocampus. This revealed changes, both increases and decreases, in the levels of several. The two other studies published around the same time focussed on single microRNAs. One of the teams looked at miR-132, finding that it was strongly up-regulated by seizures in rodents.[Reference Nudelman, DiRocco, Lambert, Garelick, Le and Nathanson225] The other study, led by wife-and-husband team Eleonora Aronica and Jan Gorter based at the University of Amsterdam, looked at miR-146a, which immunologists had recently found to coordinate inflammatory responses.[Reference Aronica, Fluiter, Iyer, Zurolo, Vreijling and van Vliet226] They found higher levels of miR-146a in the hippocampus from patients with TLE and showed that the same increase occurred in the same brain region when epilepsy was triggered in rats. In contrast to the very early and rapid rise in miR-132 that followed seizures, levels of miR-146a increased more gradually. The Dutch group took the important step of looking at what cell type was making miR-146a. It turned out to be astrocytes. In addition to hoovering up released neurotransmitter and generally contributing to the optimal environment surrounding synapses, astrocytes can mount powerful inflammatory responses after tissue injury. It appeared that miR-146a was a regulator of the inflammatory response in epilepsy, although, without manipulating the microRNA, it remained unknown whether this promoted or opposed neuroinflammation.
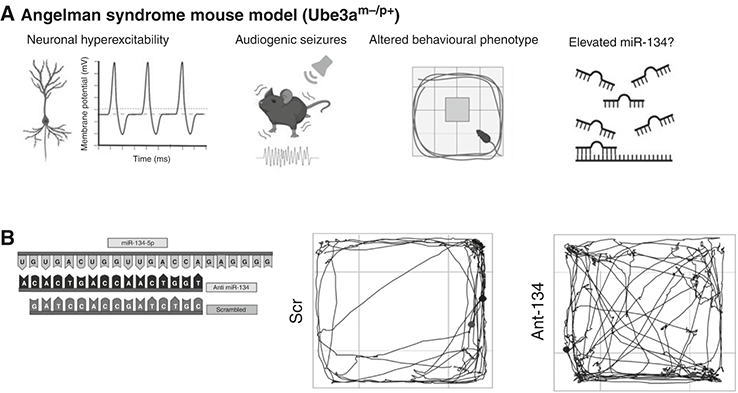
At the time these studies came out, we were sitting on our own data on microRNAs. We had used a microRNA screening kit to measure 380 different microRNAs in the hippocampus of mice given kainic acid, finding increases and decreases in the levels of 23 different microRNAs. We also had some of our own human data. For several years, we had been working with neurologist Norman Delanty at Beaumont Hospital in Dublin, a major centre in Ireland for the treatment of complex, drug-resistant epilepsy. The hospital specialises in the surgical treatment of epilepsy, procedures that were performed by neurosurgeon Donncha O’Brien. With the agreement of chief pathologist Michael Farrell, we worked to build up a collection of brain tissue samples donated after surgery by TLE patients. Then, using small pieces surplus to pathology’s needs, we extracted RNA and measured levels of microRNAs, comparing these to levels found in autopsy samples donated to a brain bank. The levels in the human samples matched many of the changes we had found in the mice. We were about to publish this when the three other studies appeared. This put the brakes on our plans. We wouldn’t be the first to publish on microRNAs and epilepsy, so the impact of reporting what we had found would be diminished. We decided to be patient and to add something more. Something no one else had tried: test whether altering one of the microRNAs would affect epilepsy. The ability to do this, to target a microRNA in a seizure model, was possible using antimirs. The main approach, at least for up-regulated microRNAs, was to inject an antimir into the brain of a lab mouse, engaging and blocking the targeted microRNA from working. We weren’t chemists so we used a company that could synthesise a microRNA inhibitor for us. We just had to tell it which one we were interested in and it sent us a batch of the antimir. We actually picked antimirs to two microRNAs, but it was the results of testing the antimir to miR-134 that would change the direction and indeed shape the future focus of my lab for the next decade.
Blocking miR-134 Prevents Seizures
We met miR-134 in Chapter 5. There were several reasons to go after this microRNA. The link to neuronal microstructure was attractive because spines are conduits of excitatory neurotransmission and changes to dendritic structure were present in resected brain tissue from TLE patients. Our first job was to figure out how much of the antimir we would need to use. No one had inhibited this specific microRNA in the brain so we ran some pilot tests, eventually finding that injection of 0.12 nanomoles was sufficient to inhibit the microRNA (there are 6 × 1023 molecules in a mole, so this is about 10 billion times less). Having got the dose right, we checked how long the antimir effects lasted before wearing off. We found that the microRNA inhibitor had effects lasting at least one month. This prolonged duration of action was very exciting since it might allow us to generate long-lasting effects on the epileptic brain. We then designed two experiments. First, we would test what happened when we lowered the amount of miR-134 in the brain before giving kainic acid to mice. This would tell us if the microRNA controlled the excitability in the normal brain. The second experiment was the more exciting and ambitious. What if we inhibited miR-134 after we had initiated the epileptogenic process? Could we prevent epilepsy developing? Or at least reduce its severity? In both experiments, inhibiting miR-134 was protective. Mice in which we had lowered brain levels of miR-134 experienced less-severe seizures when later exposed to kainic acid.[Reference Jimenez-Mateos, Engel, Merino-Serrais, McKiernan, Tanaka and Mouri183] We also found that signs of toxicity from the kainic acid and the seizures they caused, including neurodegeneration, were reduced. The results of the second experiment were the biggest surprise. Normally, mice begin to display brief epileptic seizures within a few days of kainic acid treatment. They continue to have these seizures regularly afterwards. The mice given the miR-134 inhibitor showed hardly any epileptic seizures and none at all during the first few days of monitoring. We kept watching and recording brain activity using our miniature EEGs, wondering if the effect would wear off. It didn’t. Mice were still experiencing only rare seizures weeks and up to two months later. What we had found, it seemed, was something that could strongly suppress the development of epilepsy. But we had not fully prevented epilepsy. Perhaps we could, with some further adjustments to doses and timing of injections. To put this into context, ASMs have no such effect in preclinical models and nor do they prevent the development of epilepsy following brain injury in humans. The finding would set us on course to try to turn this into a medicine, a story we will pick up in Chapter 7.
Making Sense of the Mechanism
Over the course of the next few years, we tried to understand how the antimir might be producing these effects. We have identified three potential actions. The first, revealed by the first experiment, is an acute anti-seizure effect. This occurred when we injected the antimir, waited a day for it to take effect and then triggered seizures, which we found to be less severe than would be expected. While this was exciting, a drug that has to be given beforehand has limitations. That said, this is rather the way that epilepsy is treated currently. Patients take ASMs that lower the excitability of the brain, but they take them each day, even though an actual seizure might occur only rarely. So, although this finding did not rule out some clinical use, it was not what we were most interested in. What we most wanted to find, indeed where the majority of the focus is these days in the field of new treatments for epilepsy, is something that would modify the disease, actually change the underlying biological processes that are responsible for the disease. For example, switching certain genes off that were on, or vice versa, thus altering the connections between cells or other aspects of neuronal networks, could reduce their ability to synchronise and generate seizures. The results of our second experiment suggested that we might have an anti-epileptogenic or disease-modifying effect. The injection of the antimir resulted in far fewer seizures and this lasted for weeks. We think this is permanent because we tried to detect the presence of the antimir at the end of the experiments, weeks after the injection, and we couldn’t find it. This suggested that over a period of a few days or maybe weeks, but not longer, the antimir had changed the circuitry at some level and disrupted the network that was triggering seizures.
We found one more benefit of the antimir. Epileptic mice that received the miR-134 inhibitor had more surviving neurones in their hippocampus. The molecule seemed to be neuroprotective. Was this a direct or an indirect effect of the antimir? If the antimir reduced the severity or duration of seizures, it might spare neurones from dying, but that would be an indirect effect. We suspected that the neuroprotection in the first experiment, where we gave the antimir before the kainic acid, was mostly because we had reduced the severity of the seizures. Less seizures = less damage. But in the second experiment we had given the inhibitor after the kainic acid and we still saw neuroprotection in the brains. This made it much more likely that the inhibitor was directly neuroprotective. We decided to perform a further experiment to settle the issue. We turned to neuronal cultures and set up three plates of cells. The first neurones were exposed to kainic acid alone. As expected, a check on the cells six hours later showed that half of the neurones were dying. The second plate of cells was treated with the antimir. These antimir-treated neurones all survived. The third plate contained cells given the antimir accompanied by double-stranded RNAs designed to lower levels of Limk1 by RNAi. If the antimir was protecting the neurones by preventing miR-134’s action on Limk1, effectively de-repressing LIMK1, then down-regulating Limk1 should cause the neuroprotection to disappear. This is what we found. Virtually all the neurones that had been depleted of Limk1 alongside treatment with the antimir in the plate died. This experiment told us that the neuroprotective effect of the antimir depended on LIMK1. Presumably, blocking miR-134’s effects on Limk1 caused the neurones to receive a less-severe toxic shock when the kainic acid was applied.
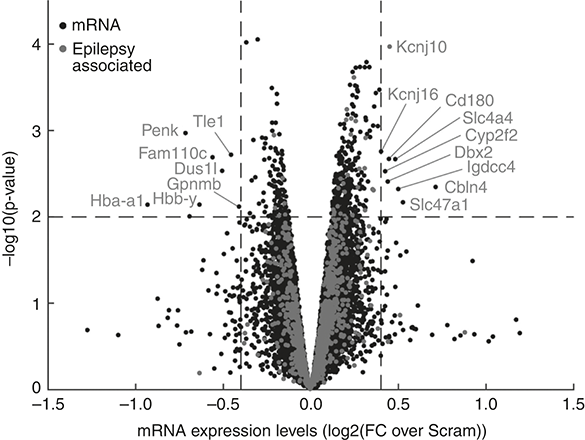
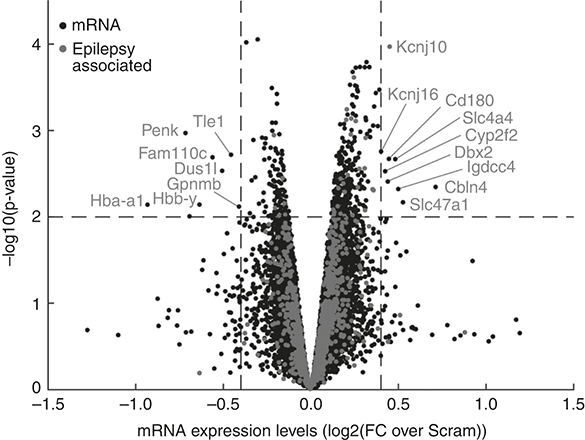
Work to understand the mechanism of protection is continuing. As with any microRNA research, the process of establishing which targets are important is complicated by the expanse of targets. I doubt that all the effects of the antimir are explained by rescuing any single target. The actions of microRNAs are context-dependent. We know that knockout of some microRNAs produces limited effects under control conditions, but upon stress or introduction of a pathology/disease context, strong phenotypes emerge. An antimir effect on a microRNA and its targets may change or be more complex as target levels and the site of action fluctuate in disease. And we must think too of the relationship between spines and epilepsy itself. Increased excitatory synaptic activity causes expansion of spines, but excessive stimulation (e.g. prolonged seizures) causes swelling and loss of spines. Severe dendritic alterations and spine loss have been reported in resected brain tissue from patients with TLE as well as genetically caused epilepsies. However, studies have also found rather normal dendritic morphology in brain samples from TLE patients despite a history of seizures, indicating that seizures alone may not always cause spine loss. Before moving on, there are two final experiments that clarify some of the mechanism of action of the antimir targeting miR-134 in the setting of epilepsy. We performed a mouse version of the cell culture experiment. That is, we lowered levels of Limk1 at the same time as giving the antimir-134, but this time in mice that had been given kainic acid and were destined to develop epilepsy. The mice, given an antisense inhibitor of Limk1 at the same time as the antimir, developed a higher number of spontaneous seizures.[Reference Reschke, Silva, Vangoor, Rosso, David and Cavanagh227] That suggested that the seizure-reducing effects of blocking miR-134 in mice depend on the antimir restoring or protecting the amount of LIMK1. However, measuring levels of transcripts in the hippocampus of mice after they were given the antimir revealed changes to levels of 39 protein-coding genes in the mice given the antimir targeting miR-134 (see Figure 6.4). Among the different genes were elevated transcripts encoding potassium channel–related proteins, a transporter for sodium and a multi-drug transporter. Many other gene changes occurred at lower levels or less consistently. These could all be part of the anti-seizure mechanism(s). This captures the challenge of assigning a single mechanism to the observed effect of inhibiting this or any other microRNA. Efforts continue to understand how altering miR-134 protects against seizures. The answer could help with discovering future therapies.
Changes to hippocampal gene expression after inhibition of miR-134
This ‘volcano’ plot shows gene expression changes in the hippocampus of kainic acid–treated mice 24 hours after inhibition of miR-134. Each dot is a unique gene transcript, with up-regulated transcripts right of centre, down-regulated to the left. The most important are specifically identified, based on statistical significance and fold change. Those genes previously associated with epilepsy are marked in lighter grey.
Disappearing Dicer
We have also measured levels of miR-134 in human brain samples. The mature sequence of miR-134 is identical between mice, rats and humans and its functions in the brain may be similar since the seed sites for several experimentally confirmed targets are also conserved (Limk1 might be an outlier here; the 3’ UTR in the human transcript, LIMK1, does not possess a canonical miR-134 seed). Tissue samples from both the hippocampus and the overlying cortex of patients operated on for treatment-resistant TLE show elevated levels of miR-134. The up-regulation of the microRNA is therefore a conserved feature of TLE. This gives me confidence that if we are to develop a drug targeting this microRNA – see Chapter 7 – we have a good chance of it working.
During our studies we have found many other microRNA changes in human TLE and have also taken a look at the broader microRNA biogenesis machinery. In most brain tissue samples we have analysed, the levels of Dicer, AGO2 and Drosha appear quite normal. But in the most damaged hippocampal tissue, where there is severe neuronal loss and gliosis, we have detected a curious change: a reduction in Dicer levels but elevation in AGO2[Reference McKiernan, Jimenez-Mateos, Bray, Engel, Brennan and Sano228] (Drosha was again unchanged). Where this was observed, we also found lower levels of many mature microRNAs. Precursor microRNAs appeared to be present at normal levels. This indicates a select failure of the microRNA system at the point of Dicer, an impairment in the ability of the remaining cells to generate the normal complement of mature microRNAs. We might speculate that the higher AGO2 level is some kind of a compensatory reaction, perhaps a way to maximise the available RISC loading of any mature microRNAs that do get made? Mice with TLE following kainic acid treatment also had reduced levels of Dicer in the most damaged part of the hippocampus, as well as elevated AGO2. The findings are evidence that the microRNA biogenesis system can be impaired in TLE. I wonder if delivering Dicer via a gene therapy approach might have therapeutic benefits in TLE? This has not been tried by anyone, as far as I know, and remains on my ‘to-do’ list.
A MicroRNA Controls Motor Seizures
In Chapter 5 we encountered structures in the central nervous system (CNS) and a microRNA that is important for control of movement: the specialised motoneurones that depend on miR-218 which are located within the spinal cord and the motor cortex of the brain. But there are other pathways in the brain that control movement. One of these systems is called the basal ganglia, which is a set of brain structures and nuclei that control, among other things, voluntary movement. One of these, located in our midbrain, is the substantia nigra and a subpart of this, called the pars compacta, is dense with dopamine-containing neurones. Signals from these neurones are required for movement and motor control. We know this, in part, because this is the region of the brain most obviously damaged in the brain of someone with Parkinson’s disease. The dopamine-producing neurones run from the substantia nigra to a brain structure called the striatum. Here, they synapse and modulate GABA-releasing inhibitory neurones. These act on and inhibit neurones in a region called the globus pallidus, which has neurones that project to another structure, the thalamus. From there, we get activation of motor pathways and initiation of movement. We are skipping some details, but the take-home message is that dopamine pathways are important for motor control. But this brain network also has a strong connection to epilepsy. Experiments show that stimulating and blocking parts of the basal ganglia circuitry can modify seizures. One of the earliest studies on this came in a 1982 paper in Science by a team at Georgetown University in the USA.[Reference Iadarola and Gale229] They showed that injecting chemicals that act like GABA into the substantia nigra blocked generalised seizures in rodents. Conversely, injecting kainic acid into the substantia nigra of rodents triggered seizures. Since that time, evidence has accumulated in labs and in the clinic for this seizure-modulating role. For example, brain imaging studies in people with epilepsy have found increased activity in the basal ganglia during generalisation of seizures, when seizure activity jumps from one place to another as the seizure spreads through the brain. While the findings have not led directly to basal ganglia–directed therapies, there are implantable medical devices that control seizures by stimulating another part of this circuit. And the amygdala, which we encountered in Chapter 5, is connected to the basal ganglia and commonly involved in the seizure circuits in TLE. Indeed, researchers apply kainic acid into the amygdala in rodents as a way to trigger seizures and model epilepsy.
Against this backdrop, Schaefer’s team explored what happened when a key brain-enriched microRNA, miR-128, was deleted from specific neurones that are innervated by the dopamine-containing neurones in the substantia nigra. Their findings, reported in Science in 2013, were a major advance in the field because they demonstrated that epilepsy could result, at least in mice, from the deletion of a single microRNA gene.[Reference Tan, Plotkin, Veno, von Schimmelmann, Feinberg and Mann230] Schaefer’s team began with a full knockout of miR-128 (Mir128−/−). Mice deficient in miR-128 were born and seemed largely normal, for a while. But they did not survive beyond two to three months of age. By watching the mice over time, they noticed that they were dying because of severe seizures. The loss of miR-128 was making the mouse brain much more excitable. They decided to narrow down the region of the brain and the cell type causing the epilepsy. This led them to a surprising finding. Deficiency of miR-128 in just a subset of the dopamine-responsive neurones of mice, those present in the striatum, resulted in the same phenotype as the full-body knockout, that is, severe motor seizures that were life-shortening. These were epileptic in nature since giving mice the drug sodium valproate, a common ASM, prevented the seizures and extended the lifespan. When they looked at the neurones of miR-128-deficient dopamine-responsive neurones, they found that they had higher spine density. Electrical recordings from the dendrite area also detected increased excitability. The study went on to show that miR-128 controlled levels of more than 150 gene transcripts. This included sodium and calcium channels and downstream signalling pathways. The team finished by demonstrating that boosting levels of miR-128 in mice protected against seizures caused by kainic acid. The study stands out as providing the first genetic evidence that a single microRNA controls the pathways that in turn control the motor component of seizures. It revealed miR-128 to be a master controller of neuronal excitability, and linked this effect to the pathways of the basal ganglia. The high-profile publication also stirred the field more generally, and more researchers became interested in what microRNAs were doing in epilepsy.
Control the Potassium, Control the Seizure
Regulating the concentration and the movement of potassium is critical to maintaining the appropriate excitability of the brain. As was covered earlier, one of the simplest ways to provoke seizure-like activity in neurones is to bathe them in a high-potassium solution. This has the effect of changing the membrane potential of the cells, de-polarising them and triggering bursts of action potentials. The human genome contains about 80 genes for components of potassium channels; even a simple animal such as C. elegans has 40 potassium channel genes. Faults in potassium channel genes give rise to a number of epilepsies, referred to as potassium channelopathies. They also give rise to some genetic causes of heart disease. But not all potassium channel variants are equally bad. Carriers of genetic variants and mutations in potassium channels display a spectrum of severities. Mutations in the KCNQ2 gene, which codes for the Kv7.2 voltage-gated potassium channel subunit, give rise to a condition called benign familial neonatal convulsions, an epilepsy syndrome that typically resolves within a few months of life. At the other end of the spectrum, variants in the KCNA2 gene, which encodes the Kv1.2 channel that contributes to the resting membrane potential and how the membrane re-polarises after action potentials, cause severe and life-threatening seizures. The importance of potassium regulation has driven several teams to look for microRNAs that target the transcripts that encode potassium channels and transporters. If dysregulated, these microRNAs could represent potential underlying causes or risks for human diseases, including epilepsy. The first study to specifically link a microRNA to the control of a potassium channel appeared in the mid-2000s in connection with potassium channels that regulate heart function. Later, in 2013, the KCNA1 transcript that encodes the Kv1.1 voltage-gated potassium channel subfamily A member 1 was shown to be targeted by miR-129.[Reference Sosanya, Huang, Cacheaux, Chen, Nguyen and Perrone-Bizzozero231] My lab joined the microRNA–potassium channel hunt briefly, as part of a collaboration with a US team based at Emory University in Atlanta. Led by Nina Gross, now at Cincinnati Children’s Hospital, they began with a specific search for microRNAs that might regulate the KCND2 transcript that encodes the Kv4.2 potassium channel, a major contributor to potassium currents via controlling dendrite excitability.[Reference Gross, Yao, Engel, Xing, Danielson and Thomas232] Although rare, KCND2 mutations can give rise to epilepsy in humans. The team found that the transcript encoding Kv4.2 was present in AGO2 pull-downs from the hippocampus of the mouse, indicating active regulation by microRNA. Searches of the 3’ UTR of the potassium channel transcript revealed a promising seed match (GGAUGC) for miR-324, a brain-enriched, moderately expressed microRNA. In further experiments, they found that manipulating miR-324 in neurones resulted in changes to levels of the Kv4.2 protein. For example, blocking miR-324 using antimirs increased the amount of the potassium channel on the surface of dendrites of hippocampal neurones. The team reasoned that this should dampen overall excitability and moved to test this in mice. Giving mice an antimir inhibitor of miR-324 resulted in higher levels of the potassium channel and, when exposed to kainic acid, they had less-severe seizures. The finding could have applications as an epilepsy treatment because delivery of the antimir into mice with chronic epilepsy resulted in about a 50 per cent reduction in spontaneous seizures. This led us and others to expand our thinking slightly. While microRNA-based targeting seems a natural fit for complex pathophysiology where you want to change many pathways, the basic mechanism by which it works could serve other purposes. You could, in theory, target a microRNA to restore levels of specific genes, including treating epilepsies where a single gene is at fault. We will return to this later in the chapter.
The search for microRNAs that regulate potassium channels has been productive. Recently, a Korean team based at Daegu Gyeongbuk Institute of Science and Technology used a slightly different search strategy to identify microRNAs that could regulate the KCNQ2 transcript.[Reference Ambros2Reference Wightman, Ha and Ruvkun3Reference Wightman, Ha and Ruvkun3] This encodes a slowly activating and deactivating potassium channel. The spectrum of epilepsy associated with mutations in this gene is wide, ranging from benign neonatal seizures to a severe epilepsy syndrome in which the brain becomes damaged by the frequent seizures (an encephalopathy). Notably, mice lacking the Kcnq2 gene do not survive long after being born, suffering fatal epilepsy. The team searched three different databases for microRNA binding sites in the 3’ UTR of the KCNQ2 transcript. This search yielded a shortlist of five microRNAs common to all three databases. The researchers went on to confirm that at least one of these, miR-106b, could regulate levels of KCNQ2. For example, increasing the level of miR-106b in neurones reduced the specific potassium channel current gated by Kv7.2, the protein product of the KCNQ2 gene, whereas inhibiting the microRNA produced the opposite effect. Specifically, inhibiting miR-106b moved neurones to a more negatively charged resting potential, making them less responsive, with delays in action potential firing. Together, these studies and ongoing work show that microRNAs regulate multiple potassium channels. By affecting their electrophysiological properties, these microRNAs fine-tune the excitability of neurones. The findings have potential applications in treating some of the genetic epilepsies and, since neuronal hyperexcitability is common to acquired epilepsies, perhaps TLE. Several questions remain to be explored. How is the correct amount of these potassium channel–regulating microRNAs set within the neuronal substructures? What system of feedback determines this? Get this wrong and it can profoundly alter neuronal excitability. So far, very few studies have linked specific transcription factors to these microRNAs and this needs to be done in the context of epilepsy. The fact that modulation of these potassium channel–targeting microRNAs can produce measurable effects on neuronal excitability strong enough to modify seizures demands carefully regulated checks and balances of the microRNAs. This is true not only for these potassium channel–regulating microRNAs, of course, and applies to the other examples in this chapter.
Calming the Flames of Inflammation
Control of neuronal structure and function is not the only way that microRNAs influence the excitability of the brain in epilepsy. Glia-produced microRNAs have important roles, by shaping how these cells respond to the injuries that can trigger epilepsy and the ongoing effects of seizures on brain tissue. Here, a lot of attention has been on microRNAs that influence inflammation. A link between inflammation and epilepsy has been known for a long time. Indeed, in the account earlier in this chapter of the treatment of the young woman with nocturnal epilepsy in the Edinburgh Medical and Surgical Journal, dated 1852, there is mention of a fever in relation to an increase in the frequency of her seizures. This was supported, upon autopsy after death, by evidence of a brain infection. Besides the shocking list of attempted remedies, the article mentions that a cold press was applied to the head, although this was unlikely to have been much help.
Epilepsy therapy has come a long way. Unfortunately for Janet Duncan, she died a decade before evidence-based research revealed that bromide had anti-seizure effects. Later we have phenobarbital, phenytoin and the modern era of ASMs. As mentioned, these mainly work by targeting ion channels and neurotransmitter systems. But some epilepsies respond to therapies that target the immune system and inflammation. These includes acute, infection-related causes and epilepsies that arise because the immune system launches an attack on one or more brain proteins, so-called autoimmune epilepsies. Autoimmune epilepsies are increasingly being found to account for new onset epilepsy. There is a growing arsenal of treatments that help treat epilepsies of an inflammatory and immune nature.[Reference Vezzani, French, Bartfai and Baram234, Reference Husari and Dubey235]
The inflammatory process serves a critical role in the brain as it does elsewhere in the body. At its simplest, the inflammatory process is how we repair damaged tissue and also how we defend against attack. There are two major divisions: the innate and the adaptive arms. The former represents the systems intrinsic to any cell that allows them to react to an event such as tissue damage, and launch the repair process. The adaptive arm comprises white blood cells such as B and T lymphocytes and their weapons, including antibodies, that recognise the agent of harm and mount a counter-attack. Both aspects are vital to health.
Brain cells have an innate capacity to activate an inflammatory response. In the healthy brain, however, many of the gene pathways that drive inflammation exist at low levels or are entirely ‘off’. But cells express sensors of damage. This includes toll-like receptors (TLRs), which comprise a series of protein structures on the surface of brain cells, each configured to react to different classes of molecule. Some respond to viral nucleic acids, others to features unique to bacteria. These systems are ancient and highly conserved, with versions present in Cnidaria, those simple animals that evolved more than half a billion years ago. When infection or damage activates TLRs, cells rapidly mobilise gene signalling pathways. One of these is called nuclear factor kappa light chain enhancer of activated B cells (NFĸB), a transcription controller that mediates key early phases of inflammation including production of releasable molecules called cytokines that coordinate the inflammatory response. One of the best understood cytokines is interleukin 1β (IL1β). We shall return to this molecule shortly.
When it comes to the brain, the wound-healing process is a bit of a tightrope. The cells of the immune system are normally excluded from brain tissue and their entry can spell trouble. Some of the molecules released by resident and incoming cells sensitise neurones and change the function of glia to reactive, hyper-vigilant states, and this can literally spark trouble for the brain. Work published in Nature Neuroscience in 2022 found that surgically removed brain tissue from treatment-resistant epilepsy patients contains significant numbers of immune cell types that are absent from control brain samples.[Reference Kumar, Lim, Hazirah, Chua, Ngoh and Poh236] Using remarkable new molecular imaging techniques, they could visualise these immune cells physically contacting resident brain cells, locking receptors on to their surface and presumably exchanging signals that direct one or other cell to specific courses of action. Since we know that some of these immune cells have cytotoxic effects, this may include killing brain cells. But we also need the inflammatory process to repair damage when it arises. A 2014 study of traumatic brain injury published in Nature showed that blocking early inflammatory signals from glial cells resulted in leaky, porous blood vessels in the brain and interfered with immune cell responses.[Reference Roth, Nayak, Atanasijevic, Koretsky, Latour and McGavern237] Early inflammatory reactions are essential for the recovery process.
A good example of the two-faced nature of inflammation is what happens to microglia. Injury to the brain rapidly changes the structure and function of microglia and this profoundly changes what they do. Work by Schaefer’s group has shown that microglia are normally continuously sensing neuronal activity, responding to changes in neuronal firing rates by complex changes in their inner molecular chemistry.[Reference Badimon, Strasburger, Ayata, Chen, Nair and Ikegami238] By coaxing microglia to fluorescence under a microscope and using powerful microscopes, it was possible to resolve new finer details of the structure of microglia. It turns out that microglia continuously extend and retract delicate processes to synapses. As neuronal activity ramps up, the probing towards synapses also increases. Microglial processes become more wedged and embedded among the neuronal circuitry. This happens because of receptors on their surface that sense ATP. Microglia also make an enzyme that breaks down ATP into adenosine, which acts to reduce the further release of neurotransmitters. A natural function of microglia appears to be to limit excessive excitation in the brain. That is, microglia locally produce a signal in response to rising neuronal activity that opposes further neurotransmitter release.
If microglia depart the synapse for any reason, the brain loses one of the endogenous mechanisms it relies on to keep things calm. Unfortunately, this is exactly what happens after a brain injury. A key job for microglia is moving to sites of damage and scavenging tissue debris. Since they must desert their posts on the synapse watch, this action comes at the expense of synaptic homeostasis. So, when there is brain tissue damage, we lose some of our natural inhibitory ‘tone’. Other experiments have shown how important microglia are to setting the excitability of brain networks. Experiments where microglia were depleted from the brain saw animals become hypersensitive to stimulation. Exposure to a normally innocuous dose of kainic acid produces powerful seizures in mice lacking microglia. Even without an external stimulus, neurones around sites of the brain where microglia were removed begin to synchronise, a hallmark of epileptic networks. It is curious that microglia seem to depend on a constant signal to stay alive. This signal comes in via a receptor called CSF1R. If you block the receptor, even for a relatively short period of time, microglia disappear from the brain. A team led by Michael Johnson at Imperial College London and UCB Pharma found that blocking the CSF1R receptor that keeps microglia alive reduces seizures in animals with pre-existing epilepsy.[Reference Srivastava, van Eyll, Godard, Mazzuferi, Delahaye-Duriez and Steenwinckel239] Targeting the receptor has also been reported to improve outcomes such as cognition in models of traumatic brain injury.
A protracted inflammatory state is strongly linked to the development of treatment-resistant epilepsy, but if we try to block inflammation and get the timing wrong, outcomes can be much worse. Anti-inflammatory drugs including steroids, immunoglobulins and small molecule inhibitors of inflammatory signalling have been used with varying degrees of success to treat childhood and infection-related epilepsies. Clarifying the causes and the players in persistent inflammatory tone in brain tissue from patients and animal models may improve the use of these drugs and lead us to new and more effective approaches. Progress is being made in how cytokines change the properties of neurotransmitter receptors and the ion channels that underlie neuronal firing. Cytokines can activate enzymes that then modify the chemistry of neurotransmitter systems. The response and the sensitivity of neurones to neurotransmitters directly change in an inflammatory environment, favouring increased excitability. Few parts of the neurotransmission system are spared. Indeed, the systems used to reabsorb or inactivate released neurotransmitters are also impaired by inflammatory cytokines. This means that neurotransmitters stay around synapses longer, producing stronger effects in some cases but leading to desensitisation in others. Cytokines also make the microvessels in the brain leakier. This provides a permissive environment that encourages and facilitates entry of immune cells. There are several mechanisms by which IL1β provokes hyperexcitability. In addition to effects on neurotransmitter components, IL1β is a potent activator of astrocytes, stimulating their expansion. This alters excitability in different ways, but a key mechanism is that astrocytes are the major repository of the enzyme that breaks down adenosine, called adenosine kinase (ADK). More ADK means faster clearing of the brain’s natural anticonvulsant. This makes it more likely that a seizure can start and, once underway, harder to stop.[Reference Boison219] Any pathways that drive IL1β are therefore critical targets for an inflammation-based approach to treating epilepsy. Recent studies on miR-146a suggest that it functions to dampen down the IL1β pathway. Consistent with this, the infusion of miR-146a into mice with epilepsy was shown to reduce the occurrence of seizures.[Reference Iori, Iyer, Ravizza, Beltrame, Paracchini and Marchini240] So, microRNA control of the IL1β system offers new opportunities for therapies for epilepsy. Or, put another way, control the IL1β system and you control the epilepsy.
The link between microRNA and IL1β caught our interest. Back in 1998, I was looking at some of the proteins known to control cell death, a family of enzymes called caspases. I was interested in finding out whether blocking these might protect neurones from dying. A few members of the caspase family did not control cell death but instead were part of the pathway by which cytokines are produced. The first member of the family, Caspase-1, was originally called interleukin-1 converting enzyme. As its name suggests, the enzyme processes the precursor form of what will become IL1β. The team I was working with at the University of Pittsburgh had access to brain samples from epilepsy patients and had developed a technique to measure the activity of these caspases. All caspases are originally made in an inactive form called pro-enzymes. They must be cleaved to rearrange and form the active enzyme. You can monitor this by measuring a reduction in the amount of the heavy pro-enzyme form relative to an accumulation of the lighter active form. Looking at Caspase-1, we noticed a strong signal at the correct weight for the active form in the brain samples from the epilepsy patients. Caspase-1 was active, indirect evidence that IL1β was being produced in the brains of people with drug-resistant TLE.[Reference Henshall, Clark, Adelson, Chen, Watkins and Simon241] Animal and later clinical studies by Annamaria Vezzani in Milan, Italy and collaborators would go on to test drugs that targeted this in the first human trials.
So, the machinery for making IL1β is sitting active in the seizure zone. Let’s think upstream now. It is often a goal to find the earliest or proximal event in a cascade and to intervene at that level to have greatest impact. How or what activates the Caspase-1 enzyme? Put another way, what lies between ATP being released, that is, the damage signal, and Caspase-1 being activated? The answer was found in the mid-1990s, much of the detective work being led by the team of Klaus Schulze-Osthoff at Tübingen University in Germany. They studied the receptors for ATP, finding that some of these contained an ion channel. These would eventually be named P2X receptors (the ‘P1’ nomenclature is reserved for the adenosine-responsive receptors). Seven members of the P2X family have been discovered in the human genome, each built from three subunits and possessing a channel down the centre that opens upon binding ATP.[Reference Khakh and North242, Reference Surprenant and North243] This triggers a pulse of sodium and calcium entry into the cell and the efflux of potassium. The seventh member, P2X7, differs in certain aspects. In particular, only high levels of ATP can activate the receptor. This means that the P2X7 receptor is probably inactive under normal conditions. When tissue injury occurs or there is a strong release of ATP, the receptor becomes activated. In this way, the P2X7 receptor is tuned to respond to damage. The German team were key in pinpointing that the P2X7 receptor lies upstream of the IL1β release pathway, showing that cells lacking P2X7 receptors but containing all the other components do not release IL1β in response to ATP. The coupling appears to occur via a mixture of effects. As the P2X7 receptor opens, potassium effluxes from the cell and calcium enters, stimulating a pulse of oxidative stress that combines to drive the pathway. There has been some debate since on the role of Caspase-1 in this and there is evidence that IL1β can be released via P2X7 receptor activation without Caspase-1. Regardless, ATP activates the P2X7 receptor and this leads to IL1β release. There is also a long-running debate about what cell types make the P2X7 receptor in the brain. It is mainly present on the surface of microglia, less on other glia, and sits poised to react if injury occurs. Several studies suggest that it is also present in neurones where it might serve to modulate neurotransmitter release.
In the late 2000s around when we started our microRNA work, Tobias Engel, a postdoctoral researcher in my team, suggested that we look at the P2X7 receptor in epilepsy. This idea was nurtured by his former colleagues in Spain where he had conducted his PhD research; these included one of the pioneers in this field, M. Teresa Miras Portugal, who sadly died in 2021. They offered us some of the toolkit they had been using to study the receptor in other conditions. They had drugs to activate and block the receptor and genetically altered cells and mice that could help prove, one way or another, if the P2X7 receptor was doing anything important. We learnt quickly that, under certain circumstances and with the right doses, we could reduce seizures in mice by blocking the receptor. Tobias, working with Eva Jimenez-Mateos who was leading the miR-134 project, was to co-discover a microRNA that limits the amount of P2X7 receptor in the brain and, more broadly, acts as an inflammation dampener.[Reference Jimenez-Mateos, Arribas-Blazquez, Sanz-Rodriguez, Concannon, Olivos-Ore and Reshcke244] The route to this discovery came from searching for ‘protective’ microRNAs. We reasoned that, rather than always looking in the damaged hippocampus, we should pay some attention to the distant but connected contralateral hippocampus that was inside the other hemisphere of the mouse brain. The two hippocampi are physically connected and we knew from recording electrical signals within the contralateral hippocampus that bursts of action potentials passed through this region during a seizure. But the contralateral hippocampus did not display signs of neurone loss or much gliosis. Perhaps some sort of protective mechanism was being engaged in the contralateral hippocampus? In this way, our thinking linked back to work on tolerance, the idea that brief or sub-threshold insults awake endogenous programmes that, if given enough time to switch on, protect the brain. Might there be microRNAs in the contralateral hippocampus that serve such functions?
When we measured responses to ATP in cells in the damage zone, the ipsilateral hippocampus, we detected strong channel activity consistent with the presence of the P2X7 receptor. But we recorded only weak responses to high-level ATP in the cells on the other side. Levels of IL1β in the contralateral hippocampus were also negligible. Was something blocking the P2X7 receptor? We decided to check if the P2X7 receptor was a microRNA target. We extracted the AGO2 complex from the contralateral hippocampus and found that it contained the P2rx7 transcript. So, microRNAs were targeting the receptor. But which ones? We used some of the usual search algorithms and found that the P2rx7 transcript had 3’ UTRs for quite a few microRNAs. We needed to narrow this down so we extracted more AGO2 from the contralateral hippocampus and this time analysed the microRNAs. We then put together a list of the ones that were there, but not in the ipsilateral equivalent, and had the required seed site for the P2rx7 transcript. At the top of the list was miR-22. We set about finding out whether this was restricting levels of P2X7 receptors.
In subsequent experiments, we learnt that blocking miR-22 caused an increase in P2X7 receptor levels in the contralateral hippocampus. This was associated with more IL1β production and increased numbers of astrocytes. Epilepsy became worse than normal, with mice given the miR-22 inhibitor having more frequent seizures. This told us that miR-22 had been constraining inflammatory signalling in the contralateral hippocampus. By blocking miR-22 we had removed a critical molecular brake on inflammation. That is, miR-22 served to limit the extent of the epileptic network, acting to restrict the focus to one side of the brain and to curb, albeit incompletely, the epilepsy. Seizures were not the only aspect affected when we blocked miR-22, however. Inhibition of miR-22 made mice more anxious and less able to recognise moved objects in maze tests. Most of the effects could be prevented by treating mice with a drug that blocks the P2X7 receptor. A final experiment looked at what happened when a mimic of miR-22 was injected close to the hippocampus. This had the effect of slightly reducing the frequency of epileptic seizures in mice.
Eva and Tobias were curious about what controlled the controller. What was increasing miR-22 levels in the contralateral hippocampus? For that matter, what controlled the levels of the P2rx7 transcript? Here we had a lead already. Our Spanish collaborators tipped us off that it might be a transcription factor called Specificity protein 1 (SP1). When we delivered SP1 into neurones, it stimulated them to make the P2rx7 mRNA. It also increased levels of miR-22. Sitting upstream of both the P2rx7 gene and the gene for miR-22 was a site for SP1 to bind,[Reference Engel, Brennan, Sanz-Rodriguez, Alves, Beamer and Watters245] a promoter, which explained why both could be controlled by SP1. So, this appeared to be a homeostatic loop. The same system that increases P2X7 receptor levels also adjusts the amount of miR-22. A checks-and-balances feedback loop that serves to control pathways. But if SP1 increases miR-22, why didn’t this stop the build-up of the P2X7 receptor in the ipsilateral hippocampus? We found that high intracellular calcium interferes with the SP1 system. Perhaps the seizures hitting the ipsilateral hippocampus open calcium-permeable NMDA receptors, whereas on the contralateral side the lower seizure intensity failed to trigger such a calcium entry and SP1 could do its job raising miR-22 levels. This system of SP1 driving miR-22 to control the amount of the P2X7 receptor appears to act as a natural brake on inflammation in experimental epilepsy. But is it relevant to what is happening in human epilepsy? We have found that levels of the P2X7 receptor are increased in brain tissue from patients who had hippocampal tissue removed for the treatment of drug-resistant epilepsy. We do not know if the miR-22 system fails to act or perhaps is simply inadequate. Perhaps P2X7 receptor antagonists, if given at the right time, could be an approach to treatment. Alternatively, we could try to supplement miR-22 levels. Interestingly, P2X7 receptor antagonists are undergoing clinical trials for some brain diseases and perhaps should be tried in epilepsy. Chapter 7 will look at some of the potential ways we might move microRNAs into the realm of medicines.
Capturing It All
Shortly after our study on miR-134 was published, a research fund was announced in Europe for large-scale epilepsy projects. I had recently met a German neurologist, Felix Rosenow, who was interested in the topic and we drew together scientists that included, among others, Stephanie Schorge at UCL; Jorgen Kjems, an expert on small RNAs at Aarhus University; and the Dutch team led by Jeroen Pasterkamp, who had also become interested in microRNAs in epilepsy. Completing the team, we had Jochen Prehn, an expert in modelling complex pathways with mathematics and founder of a systems biology centre at my institute. One of the core parts of the project, named EpimiRNA, was to create a definitive atlas of microRNA changes in experimental TLE. We and others had performed various types of microRNA profiling study, but they suffered certain limitations. First, they invariably used samples from just one model. Whether such findings would extrapolate to other models, much less human epilepsy, was uncertain. Second, the methods used to identify the microRNAs had a limitation in that they did not take account of whether or not the microRNA was active. So far, everyone had been grinding up brain tissue and extracting the microRNA. While this captures the microRNAs, it cannot tell you if they are loaded into the RISC. A portion of your ‘signal’ will be from microRNAs that are not actually doing anything at the time you took the sample. A number of studies had shown that profiling the AGO-loaded microRNAs gives a more accurate impression of what microRNAs are active at a given point in time.[Reference Flores, Kennedy, Skalsky and Cullen246] But there are downsides too. The method to extract the AGO is not completely efficient, meaning that some will always get left behind or lost, and some of the complexes may break open and their contents become dispersed. Nevertheless, the approach is an important tactic in the quest to record which microRNAs are functional. We had used this AGO2 analysis approach in our mouse studies on miR-22. We planned to scale this up and run across multiple time points in different models, sequencing all the bound microRNAs. We would document all the active microRNAs at each stage of the development of epilepsy.[Reference Veno, Reschke, Morris, Connolly, Su and Yan247]
By performing an AGO2 sequencing analysis of three different animal models of epilepsy, we would be able to triangulate the results to discover which microRNA activities were shared across all the models and which changes were model-specific. Each model uses a different method to trigger epilepsy and so we knew we would find microRNA changes that were specifically reacting to, for example, kainic acid. We were also keen to take account of possible species differences. The final experimental design used two chemical models and one electrical model of epilepsy in two different mouse strains and one rat model. Samples of the hippocampus were generated by different teams to capture what might be happening at different moments during the process by which the brain became capable of generating epileptic seizures. We included a time point on the day when the animals had their first spontaneous seizure, to get as close as possible to the moment when the brain’s circuitry finally generated a true epileptic seizure. There is no way to know the exact moment this is going to happen, so this time point is really picking up both the final period of conversion to an epileptic state and the consequences of a first epileptic seizure on the microRNA environment.
All the samples were packed and sent to Kjems and his postdoctoral researcher Morten Venø (who later started his own RNA company). They pulled out the AGO2 protein from all the samples and sequenced the bound small RNAs, eventually identifying more than a billion molecules. We had a remarkable dataset, a time-map of all the microRNAs that are active as epilepsy develops, and we had information on the relative abundance of every microRNA. This is important for reasons covered in the earlier chapters of this book. A low abundant microRNA is less likely to produce biologically important effects. With the database finished, a group of us pored over it. We were looking for three things. First, assurance that the experiment had worked properly. Before looking at what might be different in epilepsy, we wanted to be sure that it contained the expected microRNAs and in the right proportions. Were the levels of microRNAs we knew to be abundant in the brain among the highest levels in our own study? Were microRNAs that are not expressed in the brain absent? This was an important sense-check that the technique had worked. The AGO2 results fitted perfectly. When the top AGO2-loaded microRNAs were listed, we found all the usual suspects. The most abundant across the three models was miR-181a, the microRNA that had been the focus of Schuman’s study in Chapter 5. The list contained many others, including miR-128 in fourth place and miR-9 in fifth. When we looked for microRNAs made in other organs in the body, such as miR-122 made in the liver, they were absent. So, the method had worked and, importantly, appeared to be faithfully representing the microRNA landscape that was known from other work. This dataset, particularly the levels at baseline, has become an important reference because it allows us to quickly look up and check whether a particular microRNA is expressed in the brain and at what level relative to others. This can be critical for influencing decisions on whether or not to pursue a particular microRNA. You might spot a new microRNA in a screen, but if its expression is very low, it might be better to keep looking.
Next, we wanted to check some of the microRNAs that had already been linked to epilepsy. We looked first for miR-132 and miR-146a because they were the two microRNAs first linked to epilepsy and they had come up in many studies since, including in our own early screening work. A rapid increase was seen in levels of miR-132 in all three models, fitting with other teams’ findings and what would be expected for an activity-regulated microRNA. Levels of miR-146a also went up in each model, but with a delay relative to miR-132, consistent with it being produced when glia react to injury and initiate inflammation and repair processes. We then looked for other patterns across the models. We were surprised with just how similar the overall amounts of AGO2-loaded microRNAs were between the two mouse strains and the rat. As an example, the abundance in terms of reads per million on a log2 scale for miR-181a was 17.3 in the black mouse, 17.2 in the white mouse and 17.1 in the rat. For miR-26a the read counts were identical across all three rodent models, at 16.2. This reinforces how highly conserved microRNAs can be. Not only are the sequences of the mature microRNAs the same between these species but the amount of them in the brain is the same. Of course, this wasn’t true of all the microRNAs; there was some divergence between the models, particularly when we looked at lower-abundant microRNAs.
Consistent with our predictions, several microRNAs showed the same change in all three epilepsy models, whereas others changed in two and some just changed in a single model. From here it was easy to identify which microRNAs were common to all models. Each phase of the process of epileptogenesis had a set of microRNAs that changed in all three models, meaning that there is a common signature of functionally engaged microRNAs in experimental epileptogenesis. The numbers of these common-to-all microRNA varied according to the time point. The lowest overlap occurred early, one hour after the epileptogenic insult had occurred. This suggested that early reactions to brain injury are most strongly shaped by the specific method of insult. The greatest overlap was seen at 24 hours. This is still an early time point in the epileptogenic process. It suggests that some core aspects of the process affect many of the same microRNAs. What were the microRNAs among the common-to-all list? At the early phases of epileptogenesis, we found multiple microRNAs we’d come across before, including miR-132 and miR-134. Interestingly, many of the microRNAs in the early phase list were not found later. Different sets of microRNAs emerged in samples taken during the later days and weeks after the insult. Only three of the same microRNAs seen in the early phase of epilpetogenesis were still changed at the later phase. This revealed that epileptogenesis has distinct ‘waves’ of microRNA changes. What these were doing is unclear, but since the early phase was enriched with the known neuronal microRNAs, that phase appears to be enriched with microRNAs that react to the intense periods of firing that accompany the early brain injury phase. That period likely also features significant changes to neuronal morphology and even neuronal death. This settles later on, with glia changes and repair-related microRNAs becoming more obvious. Consistent with this idea, we found that several of the up-regulated microRNAs during the ‘second’ phase of epileptogenesis were indeed ones known to be enriched in glia. Thus, we can actually trace waves of cell function change by tracking the peaks and troughs of microRNAs. The microRNA changes are therefore a biomarker of the cell contributions and changes that accompany epileptogenesis.
On the day of the first spontaneous seizure, we found an almost entirely different set of microRNAs that hadn’t been active during the early phase and were also not regulated across the models in the chronic phase. This points to unique events occurring on this transition day, between a state in which spontaneous seizures do not occur to one where the epileptic circuit is now set. These are final molecular adjustments after which a new and permanent state of hyperexcitability exists. We also found a set of microRNAs shared between the models at the final time point, when spontaneous seizures had been occurring for some time. Again, this pointed to a common microRNA signature of established and, in these models, most likely drug-resistant epilepsy.
In addition to creating the dataset, we asked two more questions. First, do the same microRNAs change in the same way in human TLE? The answer was yes. Several of the microRNAs we found dysregulated in all three animal models were also changed in human epilepsy. This backed up the translational value of these models. They had generated leads relevant to human epilepsy. Second, did the changes in microRNA levels affect brain excitability? We had a set of eight microRNAs that were up-regulated in all three models at the last time point representing chronic epilepsy. Two of these were miR-132 and miR-146a, so we skipped them and focussed on the remaining six. We then deployed antimirs to block these and find out if that altered anything. Here, we were primarily looking for a change either in seizures or in something with clear relevance to the pathophysiology of epilepsy, such as neuronal death. Until this point, epilepsy researchers had hit individual microRNAs one at a time. Because of the teams of scientists that the EpimiRNA project had brought together, we could be more ambitious and decided to go for all six at the same time. After designing the targeting sequences and then testing them, we had our answer. Blocking three of the six new microRNAs reduced seizures in mice. This was a strong validation of the discovery approach and evidence of the practical applications of this type of large-scale profiling study. In addition, we learnt that the three microRNAs converged on a pathway that, among other things, controls how brain cells react to injury. Figure 6.5 shows the overlapping networks of genes related to the transforming growth factor–beta pathway, controlled by the new epilepsy-associated microRNAs. Notably, genetic variants in one of the genes in this pathway causes epilepsy in humans. By inhibiting the microRNAs that suppressed this pathway, we may have enabled the signalling system to perform more effectively. This idea was backed up by the finding that when we inhibited the pathway as a way to simulate what the microRNAs were doing, using a drug this time rather than an antimir, the seizures got worse again. The study is a good example of how microRNAs exert strong effects by converging on multiple targets in a pathway. It is also an example of how ‘team science’ approaches can take on bigger challenges that generate rich datasets for the wider research community.
Convergence of microRNAs on the TGF-beta pathway in temporal lobe epilepsy
Wiring diagram depicting the experimentally validated and predicted mRNA targets of a set of three seizure-modifying microRNAs that converge on TGF (transforming growth factor)-beta signalling, illustrating the convergence of diverse microRNA targets at the pathway level.

Treating Genetic Epilepsies by Targeting MicroRNAs
While microRNA-based targeting seems a natural solution for the complex pathophysiology of TLE where you need to hit several pathways at once, the ability to increase levels of genes by targeting their microRNA regulators has clear applications for treating other epilepsies. Genetic factors underlie, to a greater or lesser extent, many forms of epilepsy. Gene mutations can result in the effective loss of half the normal gene output. The impact this has depends on the gene. We can tolerate a drop in levels of some genes better than others. Activity of the unaffected copy of a gene may be sufficient to avoid a phenotype. If not, increasing the protein yield from the ‘healthy’ copy of the gene is a strategy to alleviate some genetic epilepsies. Since microRNAs naturally dampen expression of their target genes, blocking a microRNA that targets a gene that is faulty in epilepsy could be a way to increase, at least slightly, the level of the remaining output of the gene. Taking an example from earlier, inhibiting miR-324 in someone carrying a mutation in KCND2 might be an effective way to restore enough of the encoded dendritic potassium channel to reduce hyperexcitability. The approach of using a microRNA inhibitor to increase levels of a gene in a genetic epilepsy has, of course, limitations. The levels of the other targets of the inhibited microRNA will also change, although there may be solutions for this, which will be discussed in Chapter 7. One other limitation is that this approach will be unsuitable for genetic conditions that affect the X or Y chromosomes and genetically imprinted diseases. If the mutated gene resides on the X chromosome, then, in males, there is no other copy of that gene from which to boost gene expression by removing the microRNA repression. In fact, since one copy of the X chromosome is also silenced in female cells, the problem is common to both sexes. A similar issue arises for a set of more than 200 imprinted genes in humans. Imprinting means that one copy of the gene, either the one inherited from the mother or the one inherited from the father, is permanently inactivated.[Reference Monk, Mackay, Eggermann, Maher and Riccio248] Only one copy of the gene ever contributes to protein levels in the cell. If a mutation is present in the one version of the gene that can be transcribed, then targeting a microRNA that controls that transcript will not help. You would just end up with more of a faulty protein. There are examples of epilepsies that both are X chromosome-based and affect an imprinted gene.
Are there microRNA-based approaches we could use for genetic epilepsies, either as a precision therapy, where deployment of the microRNA inhibitor would directly restore some amount of the protein deficient in a genetic epilepsy, or because there was another link between the microRNA and the pathways altered in the genetic epilepsy? Alternatively, if a microRNA inhibitor were simply a very potent anti-seizure molecule, that might also be useful. Many of the most recently approved drugs for rare genetic epilepsies are not at all targeted (e.g. cannabidiol for Dravet syndrome), yet they provide some patients with symptom control.
We have explored a microRNA approach for Angelman syndrome. It is not a classical epilepsy but a disorder of brain development in which drug-resistant epilepsy is often a serious problem.[Reference Buiting, Williams and Horsthemke249] Angelman syndrome is caused by mutations in the gene that encodes the UBE3A protein. The main symptoms are profound cognitive impairment, problems with movement, called ataxia, and drug-resistant epilepsy. The name of the disease comes from the doctor who first fully described the condition, Harry Angelman. The UBE3A protein is highly abundant in the brain, where it serves a number of roles, including helping cells break down unwanted proteins in a similar process to the one used to break down MOV10. The UBE3A protein is particularly important for the function of inhibitory neurones, regulating the various components they need at synapses. For certain reasons not completely understood, the lack of UBE3A in Angelman syndrome disproportionately affects the cerebellum and the hippocampus. Further, UBE3A is an imprinted gene. The paternal copy is normally silenced in neurones. This means that all expression comes from the maternal copy of the gene. Curiously, the imprinting occurs only in the brain, with the gene expressed from both maternally and paternally inherited genes elsewhere in the body. Patients with Angelman syndrome possess a fault in the maternal copy of UBE3A, effectively meaning that they can’t produce any useful UBE3A protein because the paternal copy is silenced. There isn’t yet a specific treatment for Angelman syndrome, with most medicines being used to simply manage symptoms. However, gene therapy approaches are being trialled that aim to interfere with the imprinting process and allow expression of UBE3A from the healthy but normally silenced paternal copy.
We picked Angelman syndrome because of a curious molecular link to microRNA dysregulation that had been discovered in 2015. The team of Schratt who first linked miR-134 to control of dendritic spines had found that one of the RNAs transcribed from the UBE3A gene contained a series of binding sites for microRNAs.[Reference Valluy, Bicker, Aksoy-Aksel, Lackinger, Sumer and Fiore250] This is nothing unusual, of course. But what was unusual was that the UBE3A transcript in question could not generate a full-length protein. It was a so-called non-protein-coding RNA variant of UBE3A. The presence of microRNA sites was not going to serve the usual purpose of reducing the translation of the transcript into protein. What might the function of this non-coding transcript be? By manipulating the amounts of the Ube3a variant in rodent cells, the team found that it was important for synaptic structure. This in itself was a noteworthy finding because the human genome contains and generates large numbers of these types of non-coding RNAs but we still know relatively little about what they do, if anything at all. What caught their eye, and later ours, was that this unusual Ube3a transcript contained a seed site for miR-134, not only in the rodent, where most of the research was focussed, but also in the human UBE3A transcript. It also contained sites for many other microRNAs from the same cluster that miR-134 is expressed from. Could this UBE3A variant actually be working to control microRNA levels? Acting like a ‘sponge’ to keep a pool of microRNAs in check? If this were true, then in Angelman syndrome the levels of the sponge would be lower and the microRNAs that would normally be attached could be available to go off and bind to other targets. In effect, Angelman syndrome might create a state akin to what we had seen in TLE, where levels of miR-134 were higher. If this were the case, perhaps our microRNA inhibitor could help to treat the disorder? At a minimum, it might work against the epilepsy and perhaps it could correct one or two of the other core symptoms.
We set out to test this idea.[Reference Campbell, Morris, Sanfeliu, Augusto, Langa and Kesavan251] We began by measuring levels of miR-134 in brain samples from a mouse model of Angelman syndrome in which a mutation was introduced that disrupted the maternal copy of the Ube3a gene. The mice are quite a good model for Angelman syndrome as they display several hallmarks, including altered synaptic structure and function. Hippocampal levels of miR-134 were, however, normal, casting some doubt on whether the loss of the ‘sponge’ Ube3a transcript in Angelman model mice was sufficient to perturb miR-134 or other microRNA levels. We pressed on. When we checked the cerebellum, we detected a small elevation in miR-134 levels. This seemed to suggest that we were on to something after all. The cerebellum is an important structure that regulates multiple brain functions, including motor coordination. There is also evidence that the cerebellum exerts a strong influence on overall brain excitability and seizures. Esther Krook-Magnuson, now at the University of Minnesota, has performed elegant experiments showing that activating certain parts of the cerebellum can interrupt seizures in the hippocampus.[Reference Krook-Magnuson, Szabo, Armstrong, Oijala and Soltesz252] Since Angelman syndrome features both reduced mobility and epilepsy, any molecular changes in the cerebellum could be relevant. Of course, there could be other reasons why the level of miR-134 differed in the cerebellum rather than it being owing simply to loss of the microRNA ‘sponge’. For example, we know that miR-134 is activity-regulated and perhaps this part of the brain was more excitable and therefore driving more miR-134, independent of the lost sponge defect. However, a quick check of the levels of some known activity-regulated genes, those which always switch on when there is high firing of neurones, suggested that this wasn’t the cause of the elevated miR-134 in this part of the brain. The evidence for a disturbance to miR-134 in Angelman syndrome remained slim, however. We decided to take a look at the levels of the unusual Ube3a RNA. One of the reasons for this check was another study, published since the work by the Schratt team, that searched for the transcript and could only find it at extremely low levels. There was scepticism in the Angelman field that this transcript could really perturb microRNAs.[Reference Avagliano Trezza, Sonzogni, Bossuyt, Zampeta, Punt and van den Berg253] We looked for it ourselves, managing to detect it, both by sifting through published mouse and human brain data generated by other groups and by analysing our own mouse and human samples. Here, we had RNA from neurones that had been reprogrammed from blood cells from an Angelman patient. Levels of the UBE3A microRNA sponge were very low but were nevertheless detectable. So, the conclusion? The microRNA sponge is present in the brain, but perhaps not at a level that could really cause much disruption to microRNA levels. Perhaps functional studies would help resolve the debate. Since the cerebellum is a key structure in movement and locomotion, we first tested whether inhibiting miR-134 with an antimir would have any effect on movement. Mice with the Angelman mutation have a characteristic gait that is slow and they also tend to avoid open spaces, a behaviour interpreted as elevated anxiety. Treating Angelman model mice with an miR-134 inhibitor (antimir) resulted in them becoming more mobile in the test area, moving slightly more quickly and also crossing over into the centre of the field (see Figure 6.6). This suggested that at least two of the symptoms of the Angelman model mice could be corrected using this microRNA approach. However, there are other ways to interpret this data. Perhaps we had interfered with the animals’ natural and protective aversion to open spaces? Perhaps the animal is now less aware of potential dangers or is even slightly confused and thus walking into areas that it would not normally enter? Figuring out which explanation is correct is not trivial and, as a lab focussed on treating epilepsy and ill-equipped to tackle interpretation of mouse behaviours, we were not able to separate these two possibilities and so moved on to the epilepsy question. Could inhibiting miR-134 stop seizures in Angelman mice? Unfortunately, this is where the mouse fails to copy the human condition. Our mice did not develop epileptic seizures. They were, however, sensitive to loud noises. Stimuli such as rattling a cage would cause the mice to have an audiogenic seizure. These are rare in humans, but there are people who have seizures in response to certain sounds and even music. We therefore tested whether blocking miR-134 could reduce audiogenic seizures in Angelman mice. It did. Angelman mice given antimirs against miR-134 developed less-severe seizures in response to a cage rattle, and it worked in both young and older mice. This was important because there was evidence that some of the features of Angelman syndrome cannot be corrected beyond a certain age. The hyperexcitability had been reported as one of those features. But we noticed some distinct differences in how the inhibitor performed in Angelman syndrome compared to our model of TLE. In particular, the anti-seizure effect didn’t last very long. Retesting the antimir-treated Angelman mice just a week later revealed that they were no longer protected. This contrasted with the months-long effects we had seen in the TLE model. We think this is because of the enduring genetic basis for the hyperexcitability in Angelman syndrome. But it could also be around dosing. It could also be that the part of the brain driving the seizure susceptibility is not the same between these conditions. We would like to find answers to some of these questions in the future.
Reduced ataxia in Angelman mice after inhibition of miR-134
Panel A shows some of the phenotypes of Angelman model mice.Panel B shows the antimir sequences and (right) representative track plots of juvenile Angelman model mice (Ube3am−/p+) moving around a test arena. Treatment with the inhibitor of miR-134 resulted in more exploring and centre crossings compared to a scrambled version of the sequence (Scr).
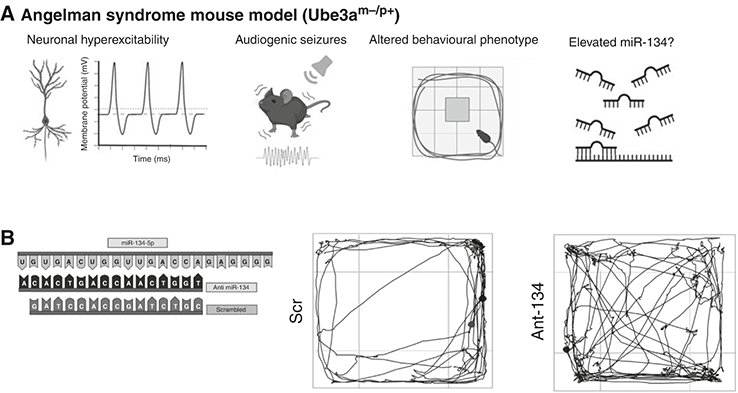
Is this approach worth pursuing as a treatment for Angelman syndrome in humans? Improvement in epilepsy and motor control would be a substantial therapeutic gain, but it would come with the need to undergo repeated injections of the antimir. It was time to look again at human samples to see if any of this would translate to patients. We did this in two ways. The first step was to see if we could modulate miR-134 in human neurones. We turned to our Angelman patient neurones and incubated them for a few days with the microRNA inhibitor. This nicely reduced the levels of miR-134 and caused a small increase in the levels of some of the known targets of miR-134. This told us that we could inhibit miR-134 in human neurones just as well as mice. We found that levels of miR-134 were no different in human Angelman neurones compared to control human neurones, questioning again the microRNA sponge hypothesis. We are still looking through some data to resolve this issue, having screened microRNAs across the mice, a set of donated brain tissue samples from Angelman patients and the human Angelman neurones. At the moment, it looks like the Angelman mouse hippocampus does express higher levels of some of the microRNAs for which binding sites existed in the Ube3a sponge transcript (but not miR-134). This does not appear to be the case for the human Angelman neurones. But does this mean that the mouse is a poor model of the human or that the human neurones we had were a poor model of the human brain? Culturing neurones and turning them into something akin to a mature brain circuit is not easy and such in vitro models lack many of the elements of the brain, including many of the non-neuronal cell types. But, actually, the human neurone microRNA profiles look similar to the human brain tissue. So, it looks like the microRNA sponge effect may be most relevant to mice. Why the species difference? We don’t know and it could be something specific to this neurodevelopmental disorder. If this divergence holds for other neurodevelopmental disorders, it will have important implications for the translatability of mouse findings for such genetic conditions in humans. The answer may also lie elsewhere. The molecular defect in our mice and our human cells was not the same. The mouse model carries an error that only prevents the production of the UBE3A protein. In contrast, the cells we used were from a patient carrying a chromosomal deletion. In these, UBE3A was lost but so were several other genes. Perhaps this molecular difference will be important in understanding why the mouse and human profiles did not align better?
Another Roll of the Dice for Dravet
Midway through the Angelman study, we became interested in another genetic epilepsy called Dravet syndrome. First described by Charlotte Dravet in an obscure French journal in 1978, Dravet syndrome is also a rare disease, affecting about 1 in every 15,000 people. The condition has three distinct phases. The first is the presentation of a severe seizure in infancy, usually accompanying a fever. Shortly after recovering, seizures return and become more severe. This continues and often worsens. Sudden death claims many infants with the gene defect. The seizures are so frequent and severe that they directly damage the developing brain, often leaving lifelong impairments in cognition and movement control. Later in life the seizures may ease, but the cognitive impairments remain. In 2001, a series of three papers in the American Journal of Clinical Genetics identified mutations in SCN1A, a gene that makes a key protein component of sodium channels, as a cause.[Reference Wallace, Scheffer, Barnett, Richards, Dibbens and Desai254, Reference Escayg, Heils, MacDonald, Haug, Sander and Meisler255, Reference Claes, Del-Favero, Ceulemans, Lagae, Van Broeckhoven and De Jonghe256] At the start of the action potential, there is a brief opening of sodium channels which causes the membrane potential to flip to positive. Without the inward sodium current there can be no action potential. But why would a reduction in the firing properties of neurones cause a predisposition to seizures? Surely the opposite would happen, that is, reduced excitability? Experiments revealed that the channel encoded by SCN1A, called NaV1.1, is particularly enriched in inhibitory neurones.[Reference Yu, Mantegazza, Westenbroek, Robbins, Kalume and Burton257] It is critical for their high firing rates. A reduction in SCN1A levels meant reduced inhibition in the brain.
We had the idea of trying our miR-134 inhibitor in Dravet syndrome. Why? By this stage, we knew that targeting miR-134 could reduce seizures in younger mice and reduce seizures in at least one genetic model. Also, the work by Gross’ team was proof-of-concept that microRNAs could be targeted to increase an ion channel in the brain. We wondered if miR-134 levels might be elevated in Dravet syndrome, since a Chinese team had found increased miR-134 levels in children with non-genetic causes of drug-resistant epilepsy. Finally, it was possible that other gene levels altered as a result of the SCN1A defect might be adjustable by a microRNA approach. Unfortunately, inhibiting miR-134 didn’t protect mice deficient in Scn1a from seizures in two different tests and we found no evidence that miR-134 was altered in the Dravet mouse brains.[Reference Gerbatin, Augusto, Morris, Campbell, Worm and Langa258] But negative findings are not always a wasted effort. The mechanisms that underlie the seizures in Dravet probably involve different neural circuits from those in TLE or Angelman syndrome. MicroRNA-based treatments need to take account of this and be tailored to the underlying molecular imbalances in an age- and aetiology-specific manner.
Targets of MicroRNAs in the Human Epileptic Brain
The first studies to profile the full complement of microRNAs in human TLE appeared in 2012: one from our lab and one from Pasterkamp’s. This provides a good picture of what is ‘up’ or ‘down’, but it doesn’t tell you which targets the microRNA is acting on. Studies have interpreted changes to target levels as driven by a change in a microRNA where the target-pair has been previously validated. But this cannot be assumed to be the case without more direct proof. In a given cell/tissue/organ, whether a microRNA binds to a specific target depends on them being co-expressed at the same time, in the same cell and, within that cell, coming into contact within a specific region of the cell. This is particularly important in the brain where we know that many microRNAs are actually shuttled out to distant dendrites to locally regulate synaptic structure and function. A microRNA could have the potential to regulate a target, but if it is not in the right place at the right time then no regulation will occur.
How do we find out what targets are controlled by microRNAs in human epilepsy? This challenge was posed at the start of the EpimiRNA project. Tackling this, Kjems, Venø and team used samples of the hippocampus from patients with treatment-resistant epilepsy and performed an experiment called individual-nucleotide resolution cross-linking and immunoprecipitation (iCLIP). The method glues RNAs to proteins, in this case the protein AGO2, the catalytic core of the microRNA silencing complex. The sequences of the RNAs can be determined and mapped against seed sequences for microRNAs. They were able to identify which mRNAs were being actively targeted by microRNAs. The result was a database of microRNA–mRNA interactions that allows anyone interested to look up the targets of any microRNA in the human epileptic hippocampus.[Reference Heiland, Connolly, Mamad, Nguyen, Kesavan and Langa259] Or, if you prefer, look up a gene of interest and find out what microRNA(s) target it. You can now ask some interesting new questions. For example, do any microRNAs target the SCN1A transcript that is lost in Dravet? This gives you a potential set of microRNAs to block to help up-regulate SCN1A levels. The first question I asked was what is bound to miR-134 in the human hippocampus from these patients? We found 99 AGO2-linked mRNAs with seeds for miR-134 in the database. This was very exciting. It proved that this microRNA was very much a ‘multi-targeting’ molecule in the human brain. But what was missing was also interesting. Some of the known miR-134 targets, including LIMK1, were not on this list. This doesn’t completely rule them out as being under miR-134 control because false negatives are always a possibility with this type of experiment, but it does give us pause for thought. Again, just how similar are microRNA targets between rodents and humans and what are the implications for trying to turn microRNAs or their inhibitors into therapeutic agents?
In addition to using this database to validate microRNA–target interactions in the human brain, the data can also be used to get a sense of the cell type making a particular microRNA. You can take the list of transcripts that were cross-linked to AGO2 and then cross-reference that list with a database of single cell sequencing data. Single cell sequencing and the microfluidics that accompany the technology make it possible to separate individual cells from a tissue and then sequence the mRNAs inside each cell. We now know what cell makes each mRNA in the human brain. If we know that a specific microRNA is targeting a specific transcript and we know what cells make that transcript, we have a good sense of what cell(s) the microRNA is working in. Taking, for example, the list of 99 mRNAs attached to miR-134 and checking it against single cell data revealed that the transcripts were expressed in either excitatory or inhibitory neurones, not glia. This is a neuronal microRNA and one probably used by both neurone subtypes.
A Single Letter Can Change Everything
In the final part of this chapter, we re-encounter the world of RNA editing. It turns out that microRNA sequences can be altered. In particular, one of the four bases, the A, can undergo a chemical modification. If that happens, the A is read as a different base. This was first shown to happen to certain protein-coding mRNAs transcripts, but it was later found to also occur to non-coding RNAs, including microRNAs. What purpose does this serve? The answer lies with increasing the repertoire, the diversity, of a given RNA.[Reference Rosenthal and Seeburg260, Reference Eisenberg and Levanon261] Most often, this results in introducing a different amino acid from the one originally encoded by the gene. The ability to make changes to any RNA transcript independently of the DNA creates flexibility and efficiency. As we will see shortly, the effects of even a single base change can be quite remarkable.
The editing of RNA is a post-transcriptional process, meaning that it relates to the insertion and/or deletion of nucleotides within an RNA molecule, as well as conversion of nucleotides. Credit for the discovery dates to 1986 when discrepancies were noted in nucleotides between the genomic sequence and the RNA sequence of a mitochondrial protein.[Reference Benne, Van den Burg, Brakenhoff, Sloof, Van Boom and Tromp262] The most common form is A-to-I editing, which is mediated by a group of enzymes called ADARs (adenosine deaminase acting on RNA). These enzymes use a molecule of water to alter the amino group in the carbon ring of adenosine.[Reference Nishikura263] This hydrolytic deamination event results in inosine in place of adenosine. Now comes the important part. Inosine is structurally very similar to guanine. The cell machinery reads an I as a G, so the original A is now behaving as a G, forming Watson–Crick pairs with cytidine instead of uracil, adenine’s normal binding partner. So, if ADARs act on an RNA, they introduce one or more inosine which would later be read as a G. When an altered base encounters the protein synthesis machinery, we can get a new amino acid in a protein. This can have dramatic effects, particularly when it is close to the active site of an enzyme or performs key gating functions. One of the best-understood examples in the brain is the editing of the GRIA2 transcript, which encodes part of the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor for the neurotransmitter glutamate. The AMPA receptor serves as the workhorse of excitatory transmission in the brain. When glutamate binds the receptor, it opens and you get a brief pulse of sodium entering the cell. This drives the cell towards firing an action potential. Unusually, the transcript coding for the protein is almost entirely edited under normal conditions in the brain. Without this editing, the AMPA receptor is too permeable to calcium. Unwanted calcium entry can trigger neurodegeneration, and mice engineered to prevent the editing from happening develop epilepsy and die prematurely. Deletion of the ADAR enzymes produces a similar result. Thus, the brain depends on RNA editing. There are other effects of A-to-I editing. For example, if it occurs close to a splicing site, this can result in changes to the inclusion or exclusion of exons. This can produce even more extensive changes to a protein. And ADARs seem to know whether and where to edit based on key sequences of nucleotides within RNAs which attract them. They also prefer double-stranded RNA. Thus, we have an elegant system in our cells for dramatically scaling up the possible outputs of a gene, creating diversity in a highly efficient manner.
Editing of microRNAs was first spotted in 2004.[Reference Luciano, Mirsky, Vendetti and Maas264] Later studies found more and more examples. The brain was a particularly rich source, with edited microRNAs showing up in different brain regions at different times.[Reference Kawahara, Megraw, Kreider, Iizasa, Valente and Hatzigeorgiou265, Reference Ekdahl, Farahani, Behm, Lagergren and Ohman266] Hundreds of RNA editing sites have now been mapped for brain microRNAs and most of the editing appears to be carried out by ADARs. But microRNAs are not read and translated into protein. So, why edit them? Editing microRNAs produces several effects. The most obvious is that editing a site in the mature microRNA can change its alignment to its targets. This could result in loss of affinity for one mRNA and gain of affinity for another. This redirection of a microRNA’s targets has been implicated in several human diseases. But it doesn’t end there. MicroRNA editing can affect the stability of the RNA, by altering how much ‘wobble’ the molecule makes. Also, RNA editing can make an RNA more or less likely to be degraded. Editing can also affect biogenesis of microRNAs, for example by altering the recognition and processing steps that are used by Drosha to process the pri-miRNA.
We suspected that microRNA editing would be different in epilepsy and became interested as the EpimiRNA project ended. Kelvin Lau, a PhD student, began by checking for the presence of ADARs in human hippocampal samples from patients with treatment-resistant TLE. Levels were quite similar to controls and, using single cell data and other techniques, he found that they were most abundant in neurones. The search for editing took advantage of a dataset of brain samples from the Amsterdam group; having finished an analysis of microRNAs, the group passed sequencing data to us to check for editing.[Reference Lau, Nguyen, Kesavan, Langa, Fanning and Brennan267] By comparing sequences in the samples to the reference genome, we were able to estimate the number of microRNAs that were edited. In total, 45 microRNAs showed some amount of editing, which varied from less than 1 per cent of the overall amount of a microRNA upwards to nearly 90 per cent. Thus, as was seen for protein-coding mRNAs, there are some microRNA that are normally heavily edited in the brain. This tells us that whatever the original use of the microRNA, it has changed. The brain has co-opted a previously evolved microRNA for a new task. So, what is that task, and does having epilepsy change that?
We were surprised when we compared patients with controls. The amounts of editing hardly differed between the groups. Despite all the disruption to normal brain function and all the changes in cell number and structure, the brain’s ability to keep on editing microRNAs seemed to be preserved. This contrasted with what we knew about microRNA abundance. Hundreds of microRNAs showed differences in their levels in human epilepsy tissue when compared to controls. The microRNA editing system seemed undisturbed. But we did find one change that was significant: miR-376a carried a nucleotide change at position 6, right in the seed, and the editing levels of this microRNA were lower in patients. Although the drop in editing was small, it could be important. First, because brain levels of this microRNA are quite high, which is important as we’ve discussed in earlier chapters. Second, miR-376a was among the most highly edited in the brain, about 85 per cent of the total amount. Would the change in editing do anything? Computer model predictions showed that it would not affect the structural stability of the microRNA but it should affect the target pool. Plugging in the unedited and edited versions of miR-376a to a target prediction algorithm revealed that the targets were almost completely different. Despite only a single base difference, the two versions of the microRNA had only 22 targets in common out of about 500 predicted. This meant that this single base had dramatically changed what the microRNA targeted. Perhaps the edited version was regulating different genes within its normal pathways. Or maybe the microRNA was regulating entirely different pathways. If that was the case, then the change in epilepsy could be quite important.
We looked at the genes and pathways under the control of the two forms of miR-376a. When plugged into a program that predicts affected pathways, the targets of the edited form included pathways involved in neurotransmission and synaptic function. Compared to the unedited form, some of the signalling pathways differed, with what seemed like a switch between neurotransmitters. This suggests that at some point the value of the original microRNA for the brain declined. By using editing, a single base change generated a ‘new’ microRNA that had a use. Again, the efficiency is striking. The cell has to make only a single base change to a single microRNA and it has a completely new tool, ready to ensure that the gene expression landscape is balanced. Now some of this had been lost in epilepsy, where the level of editing was lower. Would this have consequences?
To prove this required functional studies. We proceeded to test what happened when we lowered the editing level in neurones. This was achieved using antimirs. Reducing the edited form in a way that mimicked the situation in epilepsy resulted in dramatic changes in the levels of protein-coding mRNAs, specifically, increased expression of more than 250 genes in human neurones. This reinforced that the editing of this microRNA is a biologically important event in neurones. The types of pathway that were changed in neurones brought a further surprise. Multiple genes linked to cellular energy control were increased. Many of the transcripts encoded proteins that allow mitochondria to do their job, generating ATP for the cell. This could have the effect of altering how neurones generate energy in epilepsy. This is useful, perhaps, because the majority of the energy (fuel) used in the brain is spent just on keeping the neurones excitable. Energy is expended to run the sodium–potassium pumps and to store and release the neurotransmitters at the synapse, which are packed with mitochondria. We know from brain imaging studies that seizures produce a surge in energy and substrate demand. So, when we tweak the target pool of this microRNA, neurones might be adapting to the prevailing situation and keeping the cellular energy tank full. But could this come at a price? Sustaining higher-than-normal metabolism is unlikely to be without a downside. At a minimum, you burn up more fuel so other things such as glucose supply and oxygen need to be ramped up. This high-energy state might also affect resilience to brain insults. Indeed, down-regulation of energy-hungry pathways had been discovered to be a way the brain naturally adapts when faced with a crisis.
Back in the early 2000s, Roger Simon and colleagues at the Oregon Health and Sciences University in Portland had been looking for ways to protect the brain from stroke, such as caused by a blocked blood vessel. They marvelled that hibernating animals could tolerate prolonged periods of low blood flow in the brain without adverse effects. One of the ways the animals survive is by shutting down metabolic processes. Without a demand for energy, the brain fares pretty well with extremely low blood flow. The researchers learnt how to mimic this hibernation state by briefly interrupting the blood supply to the brain of mice. When the mice were exposed to a full stroke a few days later, the brain was almost completely protected from damage.[Reference Stenzel-Poore, Stevens, Xiong, Lessov, Harrington and Mori268] What was the forewarning event changing in the brain to make it react so differently when later hit with a stroke? It turned out that multiple energy-hungry pathways were being switched off. So, gene turn-off could provide a sustained period of protection from harm. Going back to our microRNA editing example, if we are ramping up energy for the brain to deal with seizures, could this be at the expense of neuronal well-being?
It seems that it could. Critically, the other genes that stood out as changed when we reduced the amount of edited miR-376a were linked to degenerative diseases. Pathways including Alzheimer’s, Huntington’s and amyotrophic lateral sclerosis (ALS) came up. Reducing the amount of the edited form of miR-376a appears to switch on something else, processes that could be harmful if unregulated. So, it is possible that the adaptation to provide more fuel for the hyperexcitable brain is at the expense of long-term neuronal survival. Could this cause deterioration over time? Brain imaging studies suggest that there is some brain atrophy over time in drug-resistant epilepsy.[Reference Caciagli, Bernasconi, Wiebe, Koepp, Bernasconi and Bernhardt269] Nothing like what is seen in neurodegenerative diseases, but nonetheless significant over time. This raises a final thought. Could this be turned into a therapeutic? Could we force a change to the amounts of this edited (or unedited) form of miR-376a to adjust back the altered balance and switch pathways? Perhaps raising the level of the edited form could be protective, or lowering the amount of the unedited form would trigger a compensatory rise in the edited form? I hope to explore this idea with my lab in the future.
Reflections
This chapter has covered the emergence of microRNA links to the pathophysiology of epilepsy. We’ve seen that substantial changes occur to the microRNA landscape in response to brain injuries that cause epilepsy. Models of epilepsy each display unique microRNA fingerprints in the brain, but there is also a common set of microRNAs that undergo similar changes at every stage of the process from insult to the first spontaneous seizures and on into the chronic period of epilepsy. This has generated interesting possibilities for targeting specific microRNAs as a way to interrupt or reverse that disease process. There are several microRNAs strongly linked to epilepsy, including miR-134, and there may be prospects for them as therapies. We have also seen how microRNAs make potential targets for the genetic epilepsies and neurodevelopmental disorders in which epilepsy is a major co-morbidity. Changes to microRNA levels in these models may contribute to how a gene mutation affects brain network function, excitability and behaviours. We have also begun to learn more about the microRNA system in the human brain, through analysis of tissue and modelling in iPSC-derived neurones. In many cases, the rodent models have been quite successful in generating microRNA profiles that match the human. But we have seen examples when this is not the case.
What are some of the next important questions in the area of epilepsy and microRNA? My own priorities and interests lie with learning which processes are controlled by which microRNAs in the disease setting, and applying this knowledge for therapeutic development. Which pathways are actually controlled by microRNAs in the human epileptic brain and which are not? Not all transcripts harbour suitable sites for regulation by microRNAs. Within microRNA-controlled pathways, which targets are the most regulated? What are the implications of enrichment of microRNA control for transcripts in particular pathways?
We need comprehensive cellular resolution of microRNA expression in healthy and epilepsy brain. There is some cell type–specific data on microRNAs in the human brain, but what is needed is a way to systematically map this, to learn which cells produce which microRNAs and how this changes, if at all, during the development of epilepsy and on into the chronic stage of the disease. Coupled with this, we need to understand better how a given microRNA represses a given set of targets in that cell.
Which targets are affected most (or least)? Building up this kind of knowledge will help us prioritise how therapeutic approaches might best be achieved. Going deeper into the cells, do inhibitory and excitatory neurones depend equally on the microRNA systems? Is the extent of perturbation similar between these cell types? What about cell subtypes? We can identify, either electrophysiologically or molecularly, multiple subtypes of GABA interneuron. What is the state of the microRNA system between these subtypes and does that help explain in any way the specific vulnerabilities of these cells or their particular function in epileptic networks?
Last, we need to bring some of the molecular tools that have been developed to study microRNA activity in real time into the epilepsy field. Can we see microRNA activation switching on and off in the epileptic brain? At which synapses? Does this follow or even precede an episode of seizure activity?