Refine listing
Actions for selected content:
1417033 results in Open Access
Chemical Characterization, Structural Features, and Thermal Behavior of Sodium and Hydrogen Octosilicate
-
- Journal:
- Clays and Clay Minerals / Volume 39 / Issue 5 / October 1991
- Published online by Cambridge University Press:
- 02 April 2024, pp. 490-497
-
- Article
- Export citation
Iron Substitution in Montmorillonite, Illite, and Glauconite by 57Fe Mössbauer Spectroscopy
-
- Journal:
- Clays and Clay Minerals / Volume 35 / Issue 3 / June 1987
- Published online by Cambridge University Press:
- 02 April 2024, pp. 170-176
-
- Article
- Export citation
Quantitative Assay of Minerals for Fe2+ and Fe3+ Using 1,10-Phenanthroline: III. A Rapid Photochemical Method
-
- Journal:
- Clays and Clay Minerals / Volume 36 / Issue 4 / August 1988
- Published online by Cambridge University Press:
- 02 April 2024, pp. 379-381
-
- Article
- Export citation
Technique for Transmission Electron Microscopy and X-Ray Powder Diffraction Analyses of the Same Clay Mineral Specimen
-
- Journal:
- Clays and Clay Minerals / Volume 37 / Issue 3 / June 1989
- Published online by Cambridge University Press:
- 02 April 2024, pp. 280-282
-
- Article
- Export citation
Hydrogen Positions in Dickite
-
- Journal:
- Clays and Clay Minerals / Volume 32 / Issue 6 / December 1984
- Published online by Cambridge University Press:
- 02 April 2024, pp. 483-485
-
- Article
- Export citation
Effect of Manganese on the Transformation of Ferrihydrite into Goethite and Jacobsite in Alkaline Media
-
- Journal:
- Clays and Clay Minerals / Volume 35 / Issue 1 / February 1987
- Published online by Cambridge University Press:
- 02 April 2024, pp. 11-20
-
- Article
- Export citation
Mössbauer Spectroscopic Study of the Iron Mineralogy of Post-Glacial Marine Clays
-
- Journal:
- Clays and Clay Minerals / Volume 34 / Issue 3 / June 1986
- Published online by Cambridge University Press:
- 02 April 2024, pp. 314-322
-
- Article
- Export citation
Relationship Among Derivative Spectroscopy, Color, Crystallite Dimensions, and Al Substitution of Synthetic Goethites and Hematites
-
- Journal:
- Clays and Clay Minerals / Volume 34 / Issue 6 / December 1986
- Published online by Cambridge University Press:
- 02 April 2024, pp. 625-634
-
- Article
- Export citation
Ideality of Clay Membranes in Osmotic Processes: A Review
-
- Journal:
- Clays and Clay Minerals / Volume 34 / Issue 2 / April 1986
- Published online by Cambridge University Press:
- 02 April 2024, pp. 214-223
-
- Article
- Export citation
Thermodynamic Characterization of Clay/Electrolyte Systems
-
- Journal:
- Clays and Clay Minerals / Volume 30 / Issue 4 / August 1982
- Published online by Cambridge University Press:
- 02 April 2024, pp. 291-296
-
- Article
- Export citation
Characterization of Illitization of Smectite in Bentonite Beds at Kinnekulle, Sweden
-
- Journal:
- Clays and Clay Minerals / Volume 38 / Issue 3 / June 1990
- Published online by Cambridge University Press:
- 02 April 2024, pp. 241-249
-
- Article
- Export citation
Clay in Engineering Geology, by J. E. Gillott. 2nd edition. Elsevier, Amsterdam/New York, 1987. 74 + x pages, hardbound, DFl.145.00/US$64.50 (ISBN 0-444-42758-9).
-
- Journal:
- Clays and Clay Minerals / Volume 35 / Issue 6 / December 1987
- Published online by Cambridge University Press:
- 02 April 2024, p. 477
-
- Article
- Export citation
Unit fractions with shifted prime denominators
- Part of
-
- Journal:
- Proceedings of the Royal Society of Edinburgh. Section A: Mathematics , First View
- Published online by Cambridge University Press:
- 02 April 2024, pp. 1-11
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
We prove that any positive rational number is the sum of distinct unit fractions with denominators in $\{p-1 : p\textrm { prime}\}$
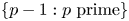 . The same conclusion holds for the set $\{p-h : p\textrm { prime}\}$
. The same conclusion holds for the set $\{p-h : p\textrm { prime}\}$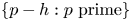 for any $h\in \mathbb {Z}\backslash \{0\}$
for any $h\in \mathbb {Z}\backslash \{0\}$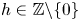 , provided a necessary congruence condition is satisfied. We also prove that this is true for any subset of the primes of relative positive density, provided a necessary congruence condition is satisfied.
, provided a necessary congruence condition is satisfied. We also prove that this is true for any subset of the primes of relative positive density, provided a necessary congruence condition is satisfied.
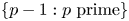
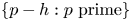
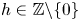
Effect of Clay Mineralogy and Aluminum and Iron Oxides on the Hydraulic Conductivity of Clay-Sand Mixtures
-
- Journal:
- Clays and Clay Minerals / Volume 33 / Issue 5 / October 1985
- Published online by Cambridge University Press:
- 02 April 2024, pp. 443-450
-
- Article
- Export citation
X-Ray Photoelectron Spectroscopic Study of Cobalt(II) and Nickel(II) Sorbed on Hectorite and Montmorillonite
-
- Journal:
- Clays and Clay Minerals / Volume 39 / Issue 1 / February 1991
- Published online by Cambridge University Press:
- 02 April 2024, pp. 22-27
-
- Article
- Export citation
Characteristics of Products from the Acid Ammonium Oxalate Treatment of Manganese Minerals
-
- Journal:
- Clays and Clay Minerals / Volume 39 / Issue 3 / June 1991
- Published online by Cambridge University Press:
- 02 April 2024, pp. 264-269
-
- Article
- Export citation
Chromate Adsorption by Kaolinite
-
- Journal:
- Clays and Clay Minerals / Volume 36 / Issue 4 / August 1988
- Published online by Cambridge University Press:
- 02 April 2024, pp. 317-326
-
- Article
- Export citation
Clay-Polymer Interactions: Summary and Perspectives
-
- Journal:
- Clays and Clay Minerals / Volume 30 / Issue 1 / February 1982
- Published online by Cambridge University Press:
- 02 April 2024, pp. 1-10
-
- Article
- Export citation
Effects of Phosphate on Iron Oxide Dissolution in Ethylenediamine-N,N,Nt’,Nt’-Tetraacetic Acid and Oxalate
-
- Journal:
- Clays and Clay Minerals / Volume 39 / Issue 3 / June 1991
- Published online by Cambridge University Press:
- 02 April 2024, pp. 324-328
-
- Article
- Export citation
Shape-Selective Sorbents Based on Clay Minerals: A Review
-
- Journal:
- Clays and Clay Minerals / Volume 37 / Issue 5 / October 1989
- Published online by Cambridge University Press:
- 02 April 2024, pp. 385-395
-
- Article
- Export citation
