Refine search
Actions for selected content:
106129 results in Materials Science
Intermediate Phases Produced During the Formation of Cadmium-Nickel Ferrites
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 1 / March 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 11-12
-
- Article
- Export citation
The X-Ray Crystallography of Tavorite from the Tip Top Pegmatite, Custer, South Dakota†
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 93-95
-
- Article
- Export citation
-
Bright apple green millimeter-sized crystals of tavorite from the Tip Top pegmatite, near Custer, South Dakota are triclinic, space group
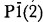 , with refined unit-cell parameters a = 5.340(2), b = 7.283(2), c = 5.110(2)Å, α = 109.29(2)°, β = 97.86(3)°, γ = 106.32(3)°, V = 174.1(3)Å3, a:b:c = 0.7332:1:0.7016, Z = 2, D(m) (suspension in methylene iodide) = 3.32(1) and D(x) = 3.33 g/cm3 (for the theoretical formula). A fully indexed X-ray powder pattern is presented. Semiquantitative electron microprobe and secondary ion mass spectroscopic analyses indicate a formula near end-member LiFe + 3(PO4)(OH). The Tip Top tavorite is biaxial positive, α = 1.795(5), β = 1.81(1), γ = 1.86(1), 2V(meas.) = 50(2)°, 2V(calc.) = 59°, XΛa ≃ 15°, YΛb ≃ 0°, and ZΛc ≃ 38°. There is no evidence for optical absorption, pleochroism or dispersion.
, with refined unit-cell parameters a = 5.340(2), b = 7.283(2), c = 5.110(2)Å, α = 109.29(2)°, β = 97.86(3)°, γ = 106.32(3)°, V = 174.1(3)Å3, a:b:c = 0.7332:1:0.7016, Z = 2, D(m) (suspension in methylene iodide) = 3.32(1) and D(x) = 3.33 g/cm3 (for the theoretical formula). A fully indexed X-ray powder pattern is presented. Semiquantitative electron microprobe and secondary ion mass spectroscopic analyses indicate a formula near end-member LiFe + 3(PO4)(OH). The Tip Top tavorite is biaxial positive, α = 1.795(5), β = 1.81(1), γ = 1.86(1), 2V(meas.) = 50(2)°, 2V(calc.) = 59°, XΛa ≃ 15°, YΛb ≃ 0°, and ZΛc ≃ 38°. There is no evidence for optical absorption, pleochroism or dispersion.
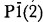 , with refined unit-cell parameters a = 5.340(2), b = 7.283(2), c = 5.110(2)Å, α = 109.29(2)°, β = 97.86(3)°, γ = 106.32(3)°, V = 174.1(3)Å
, with refined unit-cell parameters a = 5.340(2), b = 7.283(2), c = 5.110(2)Å, α = 109.29(2)°, β = 97.86(3)°, γ = 106.32(3)°, V = 174.1(3)ÅCerium Oxygen Apatite (Ce4.67[SiO4]3O) X-Ray Diffraction Pattern Revisited
-
- Journal:
- Powder Diffraction / Volume 7 / Issue 4 / December 1992
- Published online by Cambridge University Press:
- 10 January 2013, pp. 219-222
-
- Article
- Export citation
Comparison of Intensities from Fixed and Variable Divergence X-Ray Diffraction Experiments
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 2 / June 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 78-81
-
- Article
- Export citation
Cumulative Author Index
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 3 / September 1998
- Published online by Cambridge University Press:
- 10 January 2013, p. 193
-
- Article
- Export citation
Correspondents' Reports
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 3 / September 1990
- Published online by Cambridge University Press:
- 10 January 2013, p. 174
-
- Article
- Export citation
Opal, cristobalite, and tridymite: Noncrystallinity versus crystallinity, nomenclature of the silica minerals and bibliography
-
- Journal:
- Powder Diffraction / Volume 13 / Issue 1 / March 1998
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-19
-
- Article
- Export citation
PDJ volume 2 issue 3 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 3 / September 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b9
-
- Article
-
- You have access
- Export citation
Calendar of Meetings
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 2 / June 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 126-128
-
- Article
-
- You have access
- Export citation
Development of an improved XRD sample holder
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 2 / June 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 118-121
-
- Article
- Export citation
Crystal data of two thiophosphates ATi2(PS4)3 with a new structural type (A=Na, Ag)
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 2 / June 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 105-107
-
- Article
- Export citation
Correspondents Reports
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 4 / December 1989
- Published online by Cambridge University Press:
- 10 January 2013, p. 240
-
- Article
- Export citation
Correspondents Reports
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 2 / June 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 113-115
-
- Article
- Export citation
Powder X-ray data for the ammonium dioxalatotitanyl (IV) monohydrate (NH4)2TiO(C2O4)2·H2O
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 4 / December 1995
- Published online by Cambridge University Press:
- 10 January 2013, pp. 288-289
-
- Article
- Export citation
Quantitative phase analysis of α-Fe impurity in Ce2Fe17 matrix from neutron powder diffraction data
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 4 / December 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 236-239
-
- Article
- Export citation
X-ray-diffraction data of Pb2(C2O4)(NO3)2·2H2O
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 1 / March 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 7-8
-
- Article
- Export citation
On Notes for Authors
-
- Journal:
- Powder Diffraction / Volume 14 / Issue 2 / June 1999
- Published online by Cambridge University Press:
- 10 January 2013, p. 83
-
- Article
-
- You have access
- Export citation
Crystal data for [Cu(LIII)XY]·nH2O compounds [LIII=pymep, terpy; X=I, N3; Y=I, NO3, PF6; n=0,1]
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 3 / September 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 161-163
-
- Article
- Export citation
Mass Absorption Corrected X-Ray Powder Diffractograms. Part 1: Measuring Pyrite in Powdered Coals
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 1 / March 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 44-47
-
- Article
- Export citation
Commercial Announcements
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 1 / March 1990
- Published online by Cambridge University Press:
- 10 January 2013, p. 59
-
- Article
-
- You have access
- Export citation
