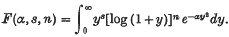Refine search
Actions for selected content:
26232 results in Theoretical Physics and Mathematical Physics
PSP volume 38 issue 3 Cover and Front matter
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 3 / July 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. f1-f2
- Print publication:
- July 1942
-
- Article
-
- You have access
- Export citation
PSP volume 38 issue 3 Cover and Back matter
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 3 / July 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. b1-b2
- Print publication:
- July 1942
-
- Article
-
- You have access
- Export citation
PSP volume 38 issue 2 Cover and Back matter
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. b1-b2
- Print publication:
- April 1942
-
- Article
-
- You have access
- Export citation
A remark on a property of a special pencil of quadrics
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 234-235
- Print publication:
- April 1942
-
- Article
- Export citation
Note on the degeneration of algebraic varieties
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 231-233
- Print publication:
- April 1942
-
- Article
- Export citation
The thermodynamic derivation of Fowler's adsorption isotherm
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 236-239
- Print publication:
- April 1942
-
- Article
- Export citation
On Lorentz invariance in the quantum theory
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 193-200
- Print publication:
- April 1942
-
- Article
- Export citation
PSP volume 38 issue 2 Cover and Front matter
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. f1-f2
- Print publication:
- April 1942
-
- Article
-
- You have access
- Export citation
On Hausdorff's methods of summability
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 166-192
- Print publication:
- April 1942
-
- Article
- Export citation
-
The aim of this paper is to give a general theory of the ‘strength’ of Hausdorff methods of summability. These methods are defined by the linear transforms
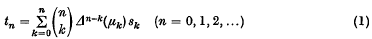
of a sequence (sk). Here the μk form a given sequence of real or complex numbers and the Δp(μk) denote their differences of order p; i.e. Δ0(μk) = μk and
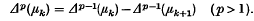
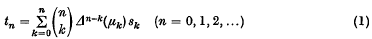
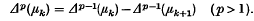
On the determination of intermolecular energies by inductive analysis
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 224-230
- Print publication:
- April 1942
-
- Article
- Export citation
-
For a direct comparison of the individual attractive and repulsive terms of an intermolecular potential determined by the inductive analysis of themodynamic data with the same terms calculated by quantal methods it is desirable to carry out the analyses, in the first approximation, with an intermolecular potential of the form ø(R) = Pe−aR − A1/R6 − A2/R8. For mathematical convenience, in place of the above expression, two potential functions,

and

are considered, the first being taken to be adequate in the range of values of R between 0 and R0 (the minimum of the potential function) and the second, in the range from R0 to ∞. By dividing the problem in this way it is possible to find substitutions which permit the integration of the classical expression for the second virial coefficients (and other appropriate thermodynamic data) directly in terms of fairly simple series in | ψ0 |, R0, a and r. Finally it is pointed out that for such simple atoms or molecules as the rare gases, oxygen, nitrogen and methane r may be taken as 0·15 throughout, which considerably simplifies the application of the method to the experimental data.


Some relations between certain methods of summation of infinite series
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 144-164
- Print publication:
- April 1942
-
- Article
- Export citation
-
Let f(t) be defined and measurable for t > 0, and suppose that it has non-vanishing moment constants
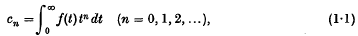
where the integrals are Cauchy-Lebesgue integrals,
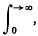 or possibly
or possibly 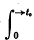 if f(t) is null for t > t0 but not for t < t0 (a convention to which we adhere throughout).
if f(t) is null for t > t0 but not for t < t0 (a convention to which we adhere throughout).
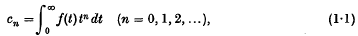
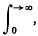 or possibly
or possibly 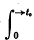 if
if A note on k-connexes
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 129-143
- Print publication:
- April 1942
-
- Article
- Export citation
-
The expression
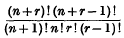
for the number of linearly independent line complexes of degree n in space of r dimensions was derived by Sisam as a particular case in the determination of the number of linearly independent hyperconnexes of given degrees. The purpose of this note is to generalize Sisam's result by determining the number of linearly independent algebraic forms of a special type to which we give the name k-connex.
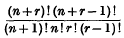
Two-centre integrals occurring in the theory of molecular structure
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 210-223
- Print publication:
- April 1942
-
- Article
- Export citation
On Lorentz invariance in the quantum theory. II
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 2 / April 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 201-209
- Print publication:
- April 1942
-
- Article
- Export citation
Relations between concentrated sets and sets possessing property C
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 1 / January 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 20-23
- Print publication:
- January 1942
-
- Article
- Export citation
On the stability of crystal lattices IX. Covariant theory of lattice deformations and the stability of some hexagonal lattices
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 1 / January 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 82-99
- Print publication:
- January 1942
-
- Article
- Export citation
PSP volume 38 issue 1 Cover and Front matter
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 1 / January 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. f1-f2
- Print publication:
- January 1942
-
- Article
-
- You have access
- Export citation
A theorem on s-dimensional measure of sets of points
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 1 / January 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 24-27
- Print publication:
- January 1942
-
- Article
- Export citation
On the stability of crystal lattices VIII. Stability of rhombohedral Bravais lattices*
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 1 / January 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 67-81
- Print publication:
- January 1942
-
- Article
- Export citation
Expansions for a particular class of exponential-logarithmic integrals
-
- Journal:
- Mathematical Proceedings of the Cambridge Philosophical Society / Volume 38 / Issue 1 / January 1942
- Published online by Cambridge University Press:
- 24 October 2008, pp. 34-39
- Print publication:
- January 1942
-
- Article
- Export citation
-
In the course of the development of a special method of interpreting thermo-dynamic data in terms of a generalized force field for the mutual interactions of the molecules concerned, expansions were required for integrals of the following general type, which apparently have not hitherto been discussed. We require explicit expansions of F(α, s, n), in terms of the parameter α, for the integral values 0, 1, 2, 3, … of s and n, where