Refine listing
Actions for selected content:
1419237 results in Open Access
Confidence regained: Providence and prayer in the works of Catherine of Siena, Anne Conway and Simone Weil
-
- Journal:
- Scottish Journal of Theology / Volume 77 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 16 December 2024, pp. 346-361
- Print publication:
- November 2024
-
- Article
- Export citation
Algebraic and geometric definitions of the cross product: the link
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 573 / November 2024
- Published online by Cambridge University Press:
- 12 November 2024, pp. 470-477
- Print publication:
- November 2024
-
- Article
- Export citation
-
Given two vectors
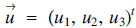 $\overrightarrow u= {({u_1},\,\,{u_2},\,{u_3})^t}$ and
$\overrightarrow u= {({u_1},\,\,{u_2},\,{u_3})^t}$ and 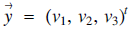 $\overrightarrow y= {({v_1},\,{v_2},\,{v_3})^t}$ in
$\overrightarrow y= {({v_1},\,{v_2},\,{v_3})^t}$ in 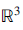 ${{\mathcal{R}}^3}$, the cross product
${{\mathcal{R}}^3}$, the cross product 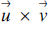 $\overrightarrow u\times \overrightarrow v $is defined as follows (see [1] or [2]):
$\overrightarrow u\times \overrightarrow v $is defined as follows (see [1] or [2]):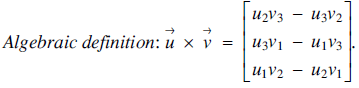 $$Algebraic{\rm{ }}definition{\rm{:}}\,\,\overrightarrow u\times \overrightarrow v \, = \,\left[ {\matrix{ {{u_2}{v_3}} \hfill &-\hfill & {{u_3}{v_2}} \hfill\cr{{u_3}{v_1}} \hfill &-\hfill & {{u_1}{v_3}} \hfill\cr{{u_1}{v_2}} \hfill &-\hfill & {{u_2}{v_1}} \hfill\cr} } \right].$$
$$Algebraic{\rm{ }}definition{\rm{:}}\,\,\overrightarrow u\times \overrightarrow v \, = \,\left[ {\matrix{ {{u_2}{v_3}} \hfill &-\hfill & {{u_3}{v_2}} \hfill\cr{{u_3}{v_1}} \hfill &-\hfill & {{u_1}{v_3}} \hfill\cr{{u_1}{v_2}} \hfill &-\hfill & {{u_2}{v_1}} \hfill\cr} } \right].$$
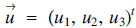
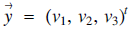
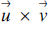
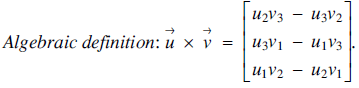
Introduction to proofs and proof strategies by Shay Fuchs, pp 349, £34.99 (paper), ISBN 978-1-00909-628-7, Cambridge University Press (2023)
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 573 / November 2024
- Published online by Cambridge University Press:
- 12 November 2024, pp. 559-560
- Print publication:
- November 2024
-
- Article
- Export citation
Lives and afterlives: The Hiberno-Latin Patrician tradition, 650–1100. By Elizabeth Dawson. Pp 179. Turnhout: Brepols. 2023. 179pp. €70 paperback.
-
- Journal:
- Irish Historical Studies / Volume 48 / Issue 174 / November 2024
- Published online by Cambridge University Press:
- 13 May 2025, pp. 388-389
- Print publication:
- November 2024
-
- Article
- Export citation
BETWEEN SAHARA AND SEA. AFRICA IN THE ROMAN EMPIRE By David J. Mattingly. Jerome Lectures Twenty-Sixth Series. University of Michigan Press, Ann Arbor, 2023. ISBN 9780472133451, pp. 744, 131 colour and black-and-white figs. Price: $44.95 (hardback)
-
- Journal:
- Libyan Studies / Volume 55 / November 2024
- Published online by Cambridge University Press:
- 16 December 2024, pp. 168-171
- Print publication:
- November 2024
-
- Article
- Export citation
Garden Kun Opera and Cultural Tourism
-
- Journal:
- New Theatre Quarterly / Volume 40 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 10 January 2025, pp. 361-371
- Print publication:
- November 2024
-
- Article
- Export citation
III. INSCRIPTIONS
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Politics as Worship: Righteous Activism and the Egyptian Muslim Brothers. Sumita Pahwa (Syracuse, NY: Syracuse University Press, 2023). Pp. 296. $39.95 paper. ISBN: 9780815638247
-
- Journal:
- International Journal of Middle East Studies / Volume 56 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 06 January 2025, pp. 710-712
- Print publication:
- November 2024
-
- Article
- Export citation
Duality of Responsibility in International Law: The Individual, the State, and International Crimes. By Thomas Weatherall. Leiden: Brill Nijhoff, 2022. 367 + xxxiv pages.
-
- Journal:
- Canadian Yearbook of International Law / Annuaire canadien de droit international / Volume 61 / November 2024
- Published online by Cambridge University Press:
- 16 December 2024, pp. 517-522
- Print publication:
- November 2024
-
- Article
- Export citation
The Americanization of the Apocalypse: creating America's own Bible. By Donald Harman Akenson. Pp xvi, 502. New York: Oxford University Press. 2023. £89 hardback.
-
- Journal:
- Irish Historical Studies / Volume 48 / Issue 174 / November 2024
- Published online by Cambridge University Press:
- 13 May 2025, pp. 398-399
- Print publication:
- November 2024
-
- Article
- Export citation
Evil and the Demonic
-
- Journal:
- New Blackfriars / Volume 105 / Issue 6 / November 2024
- Published online by Cambridge University Press:
- 18 November 2024, pp. 651-667
- Print publication:
- November 2024
-
- Article
- Export citation
BRI volume 55 Cover and Back matter
-
- Article
-
- You have access
- Export citation
Development and validation of a novel HPLC-PDA method for the detection of preservatives in milk
-
- Journal:
- Journal of Dairy Research / Volume 91 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 06 March 2025, pp. 465-470
- Print publication:
- November 2024
-
- Article
- Export citation
IPO volume 54 issue 3 Cover and Back matter
-
- Journal:
- Italian Political Science Review / Rivista Italiana di Scienza Politica / Volume 54 / Issue 3 / November 2024
- Published online by Cambridge University Press:
- 14 January 2025, pp. b1-b2
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
T. P. WISEMAN, CATULLAN QUESTIONS REVISITED. Cambridge and New York: Cambridge University Press, 2023. Pp. x + 176: illus., maps. isbn 9781009235747 (hbk), £75.00.
-
- Journal:
- The Journal of Roman Studies / Volume 114 / November 2024
- Published online by Cambridge University Press:
- 14 November 2024, pp. 174-175
- Print publication:
- November 2024
-
- Article
- Export citation
DAR volume 91 issue 4 Cover and Back matter
-
- Journal:
- Journal of Dairy Research / Volume 91 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 19 May 2025, pp. b1-b2
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
Sustaining Criminal Governance with Fear: The Use of Extra-lethal Violence to Regulate Community Life
-
- Journal:
- Journal of Latin American Studies / Volume 56 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 12 March 2025, pp. 656-682
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
