Refine listing
Actions for selected content:
1416804 results in Open Access
It's milk, Jim, but not as we know it!
-
- Journal:
- Journal of Dairy Research / Volume 91 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 19 May 2025, pp. 371-372
- Print publication:
- November 2024
-
- Article
-
- You have access
- HTML
- Export citation
Legacies and Legalities: Bequests of Land to Ecclesiastical Institutions in England c. 1180–1300
-
- Journal:
- Law and History Review / Volume 42 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 27 January 2025, pp. 691-712
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
JHS volume 144 Cover and Front matter
-
- Journal:
- The Journal of Hellenic Studies / Volume 144 / November 2024
- Published online by Cambridge University Press:
- 20 December 2024, pp. f1-f11
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
Another generalisation of Stewart's Theorem
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 573 / November 2024
- Published online by Cambridge University Press:
- 12 November 2024, pp. 478-483
- Print publication:
- November 2024
-
- Article
- Export citation
-
The aim of this Article is to present a natural generalisation of a well-known result from plane geometry, called Stewart's Theorem. We will present our result in the Euclidean space
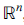 , but we will prove it only for n = 3. Since its proof for n > 3 is involved, our intention is to give the details in another Article. A search in the relative literature returns many articles which generalise Stewart's Theorem in different directions. We first refer the reader to [1], where a collection of generalisations of Stewart's theorem is presented. In [2], two results are given, which describe a relation between k points in the Euclidean space and their weighted average. If we apply these theorems to the plane, we straightforwardly get Stewart's Theorem. Another result is given in [3], which is also reproduced in [1]. This result considers n + 1 points in which belong to a hyperplane U of dimension n − 1 and another point A ∉ U , and presents an interesting relation that combines distances between each of these points and A, hypervolumes of simplices and powers of points with respect to hyperspheres. Another relative result which involves convex quadrilaterals on the plane is proved in [4]. Lastly, in [5], a generalisation of [4] is proved for 2k points in , and a relation is found between distances of these points and the distance of two corresponding averages (of these points). These articles support the fact that there can be many interesting generalisations of Stewart's Theorem in different directions.
, but we will prove it only for n = 3. Since its proof for n > 3 is involved, our intention is to give the details in another Article. A search in the relative literature returns many articles which generalise Stewart's Theorem in different directions. We first refer the reader to [1], where a collection of generalisations of Stewart's theorem is presented. In [2], two results are given, which describe a relation between k points in the Euclidean space and their weighted average. If we apply these theorems to the plane, we straightforwardly get Stewart's Theorem. Another result is given in [3], which is also reproduced in [1]. This result considers n + 1 points in which belong to a hyperplane U of dimension n − 1 and another point A ∉ U , and presents an interesting relation that combines distances between each of these points and A, hypervolumes of simplices and powers of points with respect to hyperspheres. Another relative result which involves convex quadrilaterals on the plane is proved in [4]. Lastly, in [5], a generalisation of [4] is proved for 2k points in , and a relation is found between distances of these points and the distance of two corresponding averages (of these points). These articles support the fact that there can be many interesting generalisations of Stewart's Theorem in different directions.
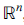 , but we will prove it only for
, but we will prove it only for Feeling the rules: Historical and contemporary perspectives on emotional norms and social distinction
-
- Journal:
- Social Science History / Volume 48 / Issue 4 / Winter 2024
- Published online by Cambridge University Press:
- 11 December 2024, pp. 603-619
- Print publication:
- Winter 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Reframing Providence: New Perspectives from Aquinas on the Divine Action Debate, by Simon Maria Kopf, Oxford University Press, 2023, pp. 320, £97, hbk
-
- Journal:
- New Blackfriars / Volume 105 / Issue 6 / November 2024
- Published online by Cambridge University Press:
- 05 December 2024, pp. 688-691
- Print publication:
- November 2024
-
- Article
- Export citation
Publications Received: (to 30 September 2024)
-
- Journal:
- Language in Society / Volume 53 / Issue 5 / November 2024
- Published online by Cambridge University Press:
- 20 December 2024, pp. 921-922
- Print publication:
- November 2024
-
- Article
- Export citation
Arianna Whelan. Reciprocity in Public International Law. Cambridge: Cambridge University Press, 2023. x + 277 pp.
-
- Journal:
- Canadian Yearbook of International Law / Annuaire canadien de droit international / Volume 61 / November 2024
- Published online by Cambridge University Press:
- 02 December 2024, pp. 513-516
- Print publication:
- November 2024
-
- Article
- Export citation
HEQ volume 64 issue 4 Cover and Front matter
-
- Journal:
- History of Education Quarterly / Volume 64 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 10 December 2024, pp. f1-f6
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
5. THE MIDLANDS
-
- Article
- Export citation
Pride, prejudice, and working-class furniture – A history of Gelsenkirchener Barock
-
- Journal:
- Social Science History / Volume 48 / Issue 4 / Winter 2024
- Published online by Cambridge University Press:
- 20 December 2024, pp. 723-749
- Print publication:
- Winter 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
L'histoire du christianisme dans la région du Djérid (sud-ouest de la Tunisie)
-
- Journal:
- Libyan Studies / Volume 55 / November 2024
- Published online by Cambridge University Press:
- 19 March 2025, pp. 143-149
- Print publication:
- November 2024
-
- Article
- Export citation
Exploring peasant and bourgeois identity: haggards, masers, sheep and spoons in later fifteenth-century County Dublin
-
- Journal:
- Irish Historical Studies / Volume 48 / Issue 174 / November 2024
- Published online by Cambridge University Press:
- 13 May 2025, pp. 199-225
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
AFR volume 94 issue 4 Cover and Back matter
-
- Article
-
- You have access
- Export citation
ILLEGALITY AND INSANITY IN TORT LAW
-
- Journal:
- The Cambridge Law Journal / Volume 83 / Issue 3 / November 2024
- Published online by Cambridge University Press:
- 07 January 2025, pp. 429-432
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Apothecia trump setae: Paratricharia belongs in the Aulaxina clade and is distant from Tricharia (lichenized Ascomycota: Gomphillaceae)
-
- Journal:
- The Lichenologist / Volume 56 / Issue 6 / November 2024
- Published online by Cambridge University Press:
- 31 December 2024, pp. 371-377
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Kunqu in Europe
-
- Journal:
- New Theatre Quarterly / Volume 40 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 10 January 2025, pp. 353-360
- Print publication:
- November 2024
-
- Article
- Export citation
Merike Blofield, Jennifer Pribble and Cecilia Giambruno, The Politics of Social Protection during Times of Crisis Cambridge University Press, 2023, pp. 77
-
- Journal:
- Journal of Latin American Studies / Volume 56 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 10 April 2025, pp. 747-749
- Print publication:
- November 2024
-
- Article
- Export citation
BJA volume 30 issue 6 Cover and Back matter
-
- Journal:
- BJPsych Advances / Volume 30 / Issue 6 / November 2024
- Published online by Cambridge University Press:
- 27 November 2024, pp. b1-b2
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
