Refine search
Actions for selected content:
106129 results in Materials Science
Identification of Crystalline Materials: Classification and Use of X-Ray Diffraction Patterns
-
- Journal:
- Powder Diffraction / Volume 1 / Issue 1 / March 1986
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-6
-
- Article
- Export citation
PDJ volume 2 issue 3 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 2 / Issue 3 / September 1987
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f19
-
- Article
-
- You have access
- Export citation
Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 4 / December 1989
- Published online by Cambridge University Press:
- 10 January 2013, p. 240
-
- Article
-
- You have access
- Export citation
Grazing excidence diffraction versus grazing incidence diffraction for strain/stress evaluation in thin films
-
- Journal:
- Powder Diffraction / Volume 15 / Issue 4 / December 2000
- Published online by Cambridge University Press:
- 10 January 2013, pp. 211-216
-
- Article
- Export citation
Données Cristallographiques sur le Trioxalato-Ferrate (III) d'Ammonium Trihydraté: (NH4)3(Fe(C2O4)3)·3H2O
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 2 / June 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 96-97
-
- Article
- Export citation
-
The trihydrated ammonium trioxalato-ferrate (III) (NH4)3(Fe(C2O4)3)·3H2O cristallizes in the monoclinic system with:
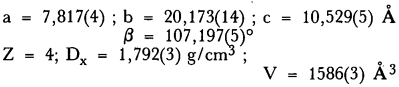
The space group is P21/c[14].
Author Index
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 4 / December 1991
- Published online by Cambridge University Press:
- 10 January 2013, p. 239
-
- Article
- Export citation
Molecular Alloys in the Series of Para-Disubstituted Benzene Derivatives. Part V. The Para-Dibromobenzene Para-Chloroiodobenzene
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 2 / June 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 85-88
-
- Article
- Export citation
Calendar of Meetings
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 1 / March 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 74-75
-
- Article
-
- You have access
- Export citation
Rietveld Analysis and Data for the Powder Diffraction File
-
- Journal:
- Powder Diffraction / Volume 10 / Issue 1 / March 1995
- Published online by Cambridge University Press:
- 10 January 2013, p. 1
-
- Article
-
- You have access
- Export citation
PDJ volume 8 issue 4 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 4 / December 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. b1-b2
-
- Article
-
- You have access
- Export citation
X-ray powder data for synthetic dolerophanite, copper(II) oxysulphate [Cu2O(SO4)]
-
- Journal:
- Powder Diffraction / Volume 9 / Issue 1 / March 1994
- Published online by Cambridge University Press:
- 10 January 2013, pp. 21-27
-
- Article
- Export citation
Computerization of the ICDD Powder Diffraction Database Critical Review of Sets 1 to 32*
-
- Journal:
- Powder Diffraction / Volume 3 / Issue 1 / March 1988
- Published online by Cambridge University Press:
- 10 January 2013, pp. 12-18
-
- Article
- Export citation
Unit-Cell Dimensions of Two-Dimensionai Clay Minerals
-
- Journal:
- Powder Diffraction / Volume 4 / Issue 1 / March 1989
- Published online by Cambridge University Press:
- 10 January 2013, pp. 19-20
-
- Article
- Export citation
Rietveld refinement of the crystal structure of α-CoSO4
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 1 / March 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. 54-56
-
- Article
- Export citation
Powder X-ray data for the solid solution [(NH4)3Al1−xFex/2Crx/2(C2O4)3] ·3H2O; 0.10≤x≤0.80
-
- Journal:
- Powder Diffraction / Volume 11 / Issue 1 / March 1996
- Published online by Cambridge University Press:
- 10 January 2013, pp. 2-4
-
- Article
- Export citation
Refinement of the Structure of MnSi by Powder Diffraction
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 4 / December 1991
- Published online by Cambridge University Press:
- 10 January 2013, pp. 194-195
-
- Article
- Export citation
Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 6 / Issue 3 / September 1991
- Published online by Cambridge University Press:
- 10 January 2013, p. 180
-
- Article
- Export citation
PDJ volume 8 issue 3 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 8 / Issue 3 / September 1993
- Published online by Cambridge University Press:
- 10 January 2013, pp. f1-f18
-
- Article
-
- You have access
- Export citation
Short Courses & Workshops
-
- Journal:
- Powder Diffraction / Volume 12 / Issue 1 / March 1997
- Published online by Cambridge University Press:
- 10 January 2013, pp. 60-62
-
- Article
- Export citation
Préparation, Identification et Radiocristallographie d'un Hétérocycle Cétonique à Intérêt Médicinal: l'Isopropyl-3, phényl-2, diaza-1,3, one-5 bicyclo [3,3,0] octène-1
-
- Journal:
- Powder Diffraction / Volume 5 / Issue 1 / March 1990
- Published online by Cambridge University Press:
- 10 January 2013, pp. 48-49
-
- Article
- Export citation
