The most prominent authigenic reaction in Holocene tuffaceous sediments at Teels Marsh, Nevada, is the hydration of rhyolitic glass by interstitial brines and the subsequent formation of phillipsite. This reaction has the form: rhyolitic glass + H2O → hydrous alkali alumninosilicate gel → phillipsite. Phillipsite is the most abundant authigenic phase in the tuffaceous sediments (>95%), analcime is the next most abundant phase, and clinoptilolite occurs as a trace mineral in the <2-mm fraction. Analcime forms by the reaction of phillipsite and Na+. Gaylussite and searlesite also are common authigenic phases at Teels Marsh. The concentration of silica in the interstitial brines is controlled by one or more of the authigenic reactions at less than 100 ppm. A stoichiometric equation for the reaction of phillipsite to analcime at Teels Marsh is:$$0.43N{a + }\, + {K_{0.43}}N{a_{0.57}}AlS{i_{3.1}}{O_{8.2}}\, \cdot \,3.2{H_2}O\, \to \,NaAlS{i_2}{O_6}\, \cdot \,{H_2}O\, + \,1.1{H_4}Si{O_4}\, + \,0.43{K + }.$$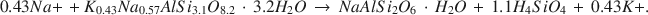
Sodium and potassium activities of brines associated with both phillipsite and analcime were used to estimate the equilibrium constant for this reaction as 3.04 × 10−5. The ΔG0 value for the reaction is +6.2 kcal/mole at 25°C and 1 atm pressure. The estimated ΔG0 value of phillipsite, using this reaction, is −1072.8 kcal/mole at 25°C and 1 atm.

