Refine listing
Actions for selected content:
1419635 results in Open Access
Human brain evolution in a Malthusian economy
-
- Journal:
- Macroeconomic Dynamics / Volume 29 / 2025
- Published online by Cambridge University Press:
- 22 November 2024, e59
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Compatibility of Kazhdan and Brauer homomorphism
- Part of
-
- Journal:
- Canadian Mathematical Bulletin / Volume 68 / Issue 1 / March 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 19-43
- Print publication:
- March 2025
-
- Article
- Export citation
-
Let G be a split connected reductive group defined over
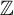 $\mathbb {Z}$. Let F and
$\mathbb {Z}$. Let F and 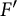 $F'$ be two non-Archimedean m-close local fields, where m is a positive integer. D. Kazhdan gave an isomorphism between the Hecke algebras
$F'$ be two non-Archimedean m-close local fields, where m is a positive integer. D. Kazhdan gave an isomorphism between the Hecke algebras 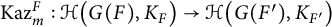 $\mathrm {Kaz}_m^F :\mathcal {H}\big (G(F),K_F\big ) \rightarrow \mathcal {H}\big (G(F'),K_{F'}\big )$, where
$\mathrm {Kaz}_m^F :\mathcal {H}\big (G(F),K_F\big ) \rightarrow \mathcal {H}\big (G(F'),K_{F'}\big )$, where 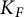 $K_F$ and
$K_F$ and  $K_{F'}$ are the mth usual congruence subgroups of
$K_{F'}$ are the mth usual congruence subgroups of  $G(F)$ and
$G(F)$ and 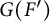 $G(F')$, respectively. On the other hand, if
$G(F')$, respectively. On the other hand, if 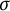 $\sigma $ is an automorphism of G of prime order l, then we have Brauer homomorphism
$\sigma $ is an automorphism of G of prime order l, then we have Brauer homomorphism 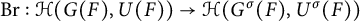 $\mathrm {Br}:\mathcal {H}(G(F),U(F))\rightarrow \mathcal {H}(G^\sigma (F),U^\sigma (F))$, where
$\mathrm {Br}:\mathcal {H}(G(F),U(F))\rightarrow \mathcal {H}(G^\sigma (F),U^\sigma (F))$, where  $U(F)$ and
$U(F)$ and 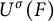 $U^\sigma (F)$ are compact open subgroups of
$U^\sigma (F)$ are compact open subgroups of 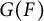 $G(F)$ and
$G(F)$ and  $G^\sigma (F),$ respectively. In this article, we study the compatibility between these two maps in the local base change setting. Further, an application of this compatibility is given in the context of linkage – which is the representation theoretic version of Brauer homomorphism.
$G^\sigma (F),$ respectively. In this article, we study the compatibility between these two maps in the local base change setting. Further, an application of this compatibility is given in the context of linkage – which is the representation theoretic version of Brauer homomorphism.
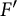
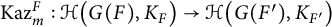
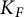


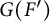
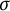
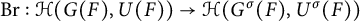

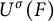
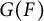

Continuing the momentum of developmental psychopathology: Lessons learned from the seminal contributions of Dante Cicchetti
-
- Journal:
- Development and Psychopathology / Volume 36 / Issue 5 / December 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 2051-2055
-
- Article
-
- You have access
- HTML
- Export citation
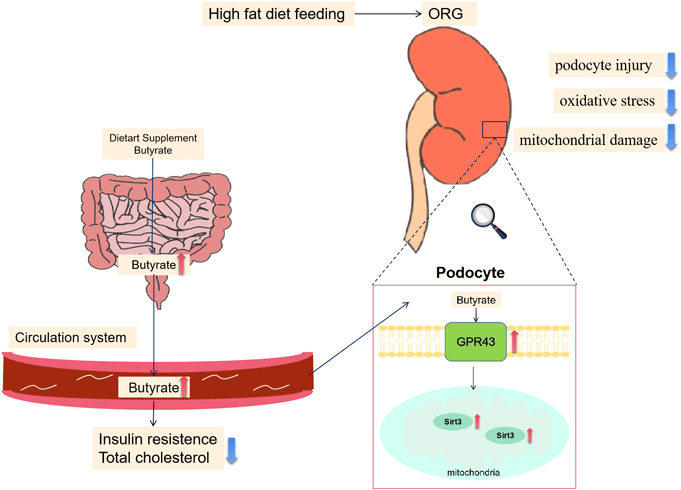
Butyrate attenuates high-fat diet-induced glomerulopathy through GPR43-Sirt3 pathway
-
- Journal:
- British Journal of Nutrition / Volume 133 / Issue 1 / 14 January 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 1-10
- Print publication:
- 14 January 2025
-
- Article
- Export citation

Asking the right questions on Rayleigh–Bénard turbulence
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 22 November 2024, F3
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The paper by Castaing et al. (J. Fluid Mech., vol. 204, 1989, pp. 1–30) on turbulent Rayleigh–Bénard convection has been one of the most impactful papers on the subject – not by giving the right and complete answers but by developing versatile concepts and by asking the right questions, namely: (i) What is the overall flow organization? (ii) What is the dependence of the Nusselt number
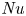 ${\textit {Nu}}$ (the dimensionless heat transport) on the Rayleigh number
${\textit {Nu}}$ (the dimensionless heat transport) on the Rayleigh number 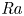 ${\textit {Ra}}$ (the thermal driving strength)? (iii) What is the ultimate state of turbulence for extremely large
${\textit {Ra}}$ (the thermal driving strength)? (iii) What is the ultimate state of turbulence for extremely large 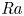 ${\textit {Ra}}$? Thanks to Castaing et al. having asked the right questions, the field has made tremendous progress over the last 35 years.
${\textit {Ra}}$? Thanks to Castaing et al. having asked the right questions, the field has made tremendous progress over the last 35 years.
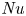
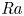
Anatomic and non-anatomic substrates in infants with two ventricles undergoing aortic arch repair
-
- Journal:
- Cardiology in the Young / Volume 35 / Issue 2 / February 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 253-260
-
- Article
- Export citation
Fence of saddle solutions of the Allen–Cahn equation in the plane
- Part of
-
- Journal:
- Proceedings of the Royal Society of Edinburgh. Section A: Mathematics , First View
- Published online by Cambridge University Press:
- 22 November 2024, pp. 1-42
-
- Article
- Export citation

Log at first sight
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 22 November 2024, F6
-
- Article
-
- You have access
- HTML
- Export citation

Ultra-fast imaging reveals
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 22 November 2024, F8
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Shipping the Color Line: Migration and Transport Policy in the British Empire, 1943–51
-
- Journal:
- Journal of British Studies / Volume 63 / Issue 4 / October 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 810-835
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Picture-Reading the Complexities of Transgender Experience
-
- Journal:
- Canadian Journal of Philosophy / Volume 53 / Issue 7-8 / October 2023
- Published online by Cambridge University Press:
- 22 November 2024, pp. 600-609
-
- Article
- Export citation
JPH volume 36 issue 4 Cover and Front matter
-
- Journal:
- Journal of Policy History / Volume 36 / Issue 4 / October 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. f1-f4
-
- Article
-
- You have access
- Export citation
Elementary quantum recursion schemes that capture quantum polylogarithmic-time computability of quantum functions
-
- Journal:
- Mathematical Structures in Computer Science / Volume 34 / Issue 7 / August 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 710-745
-
- Article
- Export citation
-
Quantum computing has been studied over the past four decades based on two computational models of quantum circuits and quantum Turing machines. To capture quantum polynomial-time computability, a new recursion-theoretic approach was taken lately by Yamakami [J. Symb. Logic 80, pp. 1546–1587, 2020] by way of recursion schematic definition, which constitutes six initial quantum functions and three construction schemes of composition, branching, and multi-qubit quantum recursion. By taking a similar approach, we look into quantum polylogarithmic-time computability and further explore the expressing power of elementary schemes designed for such quantum computation. In particular, we introduce an elementary form of the quantum recursion, called the fast quantum recursion, and formulate
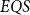 $EQS$ (elementary quantum schemes) of “elementary” quantum functions. This class
$EQS$ (elementary quantum schemes) of “elementary” quantum functions. This class 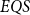 $EQS$ captures exactly quantum polylogarithmic-time computability, which forms the complexity class BQPOLYLOGTIME. We also demonstrate the separation of BQPOLYLOGTIME from NLOGTIME and PPOLYLOGTIME. As a natural extension of
$EQS$ captures exactly quantum polylogarithmic-time computability, which forms the complexity class BQPOLYLOGTIME. We also demonstrate the separation of BQPOLYLOGTIME from NLOGTIME and PPOLYLOGTIME. As a natural extension of 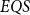 $EQS$, we further consider an algorithmic procedural scheme that implements the well-known divide-and-conquer strategy. This divide-and-conquer scheme helps compute the parity function, but the scheme cannot be realized within our system
$EQS$, we further consider an algorithmic procedural scheme that implements the well-known divide-and-conquer strategy. This divide-and-conquer scheme helps compute the parity function, but the scheme cannot be realized within our system 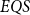 $EQS$.
$EQS$.
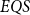
Contributors
-
- Journal:
- Ethics & International Affairs / Volume 38 / Issue 2 / Summer 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 135-136
-
- Article
- Export citation
Timed array architectures and integrated true-time delay elements for wideband millimeter-wave antenna arrays
-
- Journal:
- International Journal of Microwave and Wireless Technologies / Volume 17 / Issue 2 / March 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 190-201
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Asymptomatic Optic Disc Oedema due to Haematologic Malignancy
-
- Journal:
- Canadian Journal of Neurological Sciences / Volume 52 / Issue 5 / September 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 874-875
-
- Article
- Export citation
A criterion to detect a non-trivial homology of an invariant set of a flow in
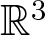 $\mathbb{R}^3$
$\mathbb{R}^3$
- Part of
-
- Journal:
- Proceedings of the Royal Society of Edinburgh. Section A: Mathematics , First View
- Published online by Cambridge University Press:
- 22 November 2024, pp. 1-31
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Consider a flow in
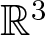 $\mathbb{R}^3$ and let K be the biggest invariant subset of some compact region of interest
$\mathbb{R}^3$ and let K be the biggest invariant subset of some compact region of interest 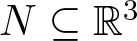 $N \subseteq \mathbb{R}^3$. The set K is often not computable, but the way the flow crosses the boundary of N can provide indirect information about it. For example, classical tools such as Ważewski’s principle or the Poincaré–Hopf theorem can be used to detect whether K is non-empty or contains rest points, respectively. We present a criterion that can establish whether K has a non-trivial homology by looking at the subset of the boundary of N along which the flow is tangent to N. We prove that the criterion is as sharp as possible with the information it uses as an input. We also show that it is algorithmically checkable.
$N \subseteq \mathbb{R}^3$. The set K is often not computable, but the way the flow crosses the boundary of N can provide indirect information about it. For example, classical tools such as Ważewski’s principle or the Poincaré–Hopf theorem can be used to detect whether K is non-empty or contains rest points, respectively. We present a criterion that can establish whether K has a non-trivial homology by looking at the subset of the boundary of N along which the flow is tangent to N. We prove that the criterion is as sharp as possible with the information it uses as an input. We also show that it is algorithmically checkable.
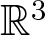
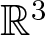
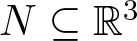
IPG volume 36 issue 10 Front matter
-
- Journal:
- International Psychogeriatrics / Volume 36 / Issue 10 / October 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. f1-f3
-
- Article
-
- You have access
- Export citation
A dual-frequency measurement setup with fully integrated SiGe-based radar sensors for the size estimation of particulate matter
-
- Journal:
- International Journal of Microwave and Wireless Technologies / Volume 17 / Issue 2 / March 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 365-372
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Adjusting to dementia as part of life: an actantial analysis of agency reconstruction following diagnosis of young onset dementia
-
- Journal:
- Ageing & Society / Volume 45 / Issue 10 / October 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 1970-1996
- Print publication:
- October 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
