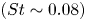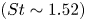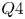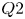Refine listing
Actions for selected content:
1419635 results in Open Access
Fotini Kondyli. Rural Communities in Late Byzantium. Resilience and Vulnerability in the Northern Aegean (New York, NY: Cambridge University Press, 2022, 290 pp., 80 illustr., 66 in colour, 20 maps, hbk, ISBN 978-1-108-84549-6)
-
- Journal:
- European Journal of Archaeology / Volume 27 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 534-538
-
- Article
- Export citation
Existence and non-existence results for a class of systems under concave-convex nonlinearities
- Part of
-
- Journal:
- Proceedings of the Edinburgh Mathematical Society / Volume 68 / Issue 1 / February 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 128-156
-
- Article
- Export citation
-
In this work, we are interested in studying the following class of problems:(𝒫λμ)
where Ω is a bounded domain in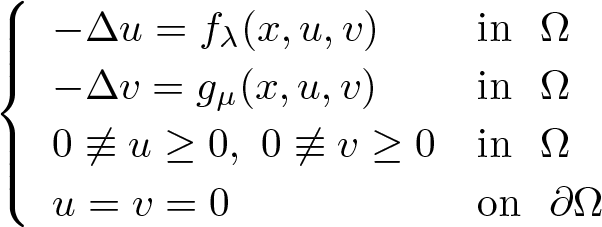 \begin{align}\left\{\begin{array}{ll}-\Delta u=f_\lambda(x,u,v)& \text{in}~~\Omega\\-\Delta v=g_\mu(x,u,v) & \text{in}~~\Omega\\0\not\equiv u\geq 0,\,\,0\not\equiv v\geq 0& \text{in}~~\Omega\\u=v=0&\text{on}~~\partial\Omega\end{array}\right.\end{align}
\begin{align}\left\{\begin{array}{ll}-\Delta u=f_\lambda(x,u,v)& \text{in}~~\Omega\\-\Delta v=g_\mu(x,u,v) & \text{in}~~\Omega\\0\not\equiv u\geq 0,\,\,0\not\equiv v\geq 0& \text{in}~~\Omega\\u=v=0&\text{on}~~\partial\Omega\end{array}\right.\end{align}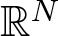 $\mathbb{R}^N$, λ > 0, µ > 0,
$\mathbb{R}^N$, λ > 0, µ > 0, 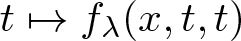 $t\mapsto f_\lambda(x,t,t)$ and
$t\mapsto f_\lambda(x,t,t)$ and 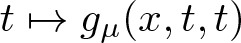 $t\mapsto g_\mu(x,t,t)$ have concave-convex type nonlinearities. We present results related to the existence and non-existence of solutions for problem
$t\mapsto g_\mu(x,t,t)$ have concave-convex type nonlinearities. We present results related to the existence and non-existence of solutions for problem  $(\mathcal{P}_{\lambda\mu})$.
$(\mathcal{P}_{\lambda\mu})$.
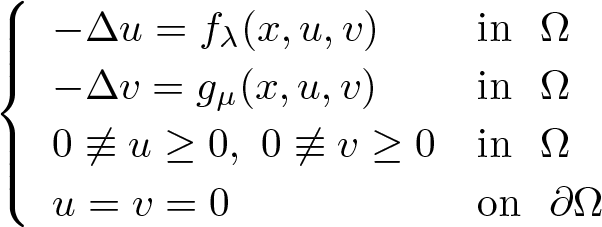
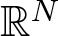
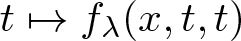
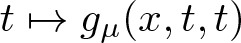

The Ethics of Special Ops: Raids, Recoveries, Reconnaissance, and Rebels, by Deane-Peter Baker, Roger Herbert, and David Whetham (Cambridge, U.K.: Cambridge University Press, 2023), 255 pp., $110 cloth, $110 eBook.
-
- Journal:
- Ethics & International Affairs / Volume 38 / Issue 2 / Summer 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 235-238
-
- Article
- Export citation
EAA volume 27 issue 4 Cover and Back matter
-
- Journal:
- European Journal of Archaeology / Volume 27 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. b1-b2
-
- Article
-
- You have access
- Export citation

Wave scattering by an isolated cyclogeostrophic vortex
-
- Journal:
- Journal of Fluid Mechanics / Volume 999 / 25 November 2024
- Published online by Cambridge University Press:
- 22 November 2024, A100
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The propagation paths of oceanic internal tides are influenced by their interactions with vortices. We examine the scattering effect that an isolated vortex in (cyclo)geostrophic balance has on a rotating shallow-water plane wave. We run a suite of simulations in which we vary the non-dimensional vorticity of the vortex,
 $Ro$, the relative scale of the vortex size to the Rossby radius of deformation,
$Ro$, the relative scale of the vortex size to the Rossby radius of deformation, 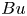 $Bu$, and the size of the vortex compared with the plane wave wavelength,
$Bu$, and the size of the vortex compared with the plane wave wavelength, 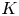 $K$. We compare the scattered wave flux pattern with ray-tracing predictions. Ray-tracing predictions are relatively insensitive to
$K$. We compare the scattered wave flux pattern with ray-tracing predictions. Ray-tracing predictions are relatively insensitive to 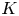 $K$ in the
$K$ in the  $1< K<4$ range we investigate; however, they generally underestimate the broad angles of the shallow-water wave scattering patterns, especially for the lower end of the
$1< K<4$ range we investigate; however, they generally underestimate the broad angles of the shallow-water wave scattering patterns, especially for the lower end of the  $K$ range. We then measure the ratio of the scattered wave energy flux to the incoming wave energy flux, denoted by
$K$ range. We then measure the ratio of the scattered wave energy flux to the incoming wave energy flux, denoted by 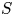 $S$, for each simulation. We find that
$S$, for each simulation. We find that 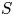 $S$ follows a power law
$S$ follows a power law 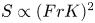 $S \propto (FrK)^2$ when
$S \propto (FrK)^2$ when 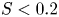 $S < 0.2$, where
$S < 0.2$, where 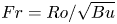 $Fr = Ro/\sqrt {Bu}$ is the Froude number. When
$Fr = Ro/\sqrt {Bu}$ is the Froude number. When 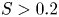 $S>0.2$, it starts plateauing.
$S>0.2$, it starts plateauing.

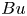
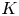
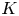


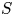
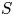
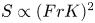
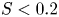
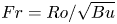
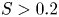

Thermal effect in shock-induced gas filtration through porous media
-
- Journal:
- Journal of Fluid Mechanics / Volume 999 / 25 November 2024
- Published online by Cambridge University Press:
- 22 November 2024, A91
-
- Article
- Export citation
Relationship Between Resilience, Emergency Response Capacity, and Occupational Stressors of New Nurse During the Re-outbreak of COVID-19 in China
-
- Journal:
- Disaster Medicine and Public Health Preparedness / Volume 18 / 2024
- Published online by Cambridge University Press:
- 22 November 2024, e285
-
- Article
- Export citation
Making the Grade: Policy Design and Effects of Information about Government Performance
-
- Journal:
- State Politics & Policy Quarterly / Volume 25 / Issue 1 / March 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 87-107
- Print publication:
- March 2025
-
- Article
- Export citation
Compact bilinear operators and paraproducts revisited
- Part of
-
- Journal:
- Canadian Mathematical Bulletin / Volume 68 / Issue 1 / March 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 44-59
- Print publication:
- March 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Sex-specific association between later circadian timing of food intake and adiposity among Chinese young adults living in real-world settings
-
- Journal:
- British Journal of Nutrition / Volume 132 / Issue 12 / 28 December 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 1629-1636
- Print publication:
- 28 December 2024
-
- Article
- Export citation
Monopoly Menace: The Rise and Fall of Cartel Capitalism in Western Europe, 1918–1957
-
- Journal:
- Enterprise & Society / Volume 25 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 22 November 2024, pp. 992-1014
- Print publication:
- December 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Sorting capsules in microfluidic devices
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 22 November 2024, F10
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Bubbling forth on thin films
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 22 November 2024, F1
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The Roles of Understanding and Belief in Prognostic Awareness
-
- Journal:
- Cambridge Quarterly of Healthcare Ethics , First View
- Published online by Cambridge University Press:
- 22 November 2024, pp. 1-9
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Multi-dimensional civic engagement of older Europeans: a latent class analysis
-
- Journal:
- Ageing & Society / Volume 45 / Issue 10 / October 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 1997-2022
- Print publication:
- October 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Care Homes in a Turbulent Era: Do They Have a Future? Pat Armstrong and Susan Braedley (eds), Edward Elgar Publishing, 2023, 184 pp., hbk £76.50, ISBN: 978 180392 581 3
-
- Journal:
- Ageing & Society / Volume 45 / Issue 2 / February 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 414-416
- Print publication:
- February 2025
-
- Article
- Export citation
Online Social Interaction for COVID-19 in Asia
-
- Journal:
- Disaster Medicine and Public Health Preparedness / Volume 18 / 2024
- Published online by Cambridge University Press:
- 22 November 2024, e286
-
- Article
- Export citation
Clinical Ethics and the Observant Jewish and Muslim Patient: Shared Theocentric Perspectives in Practice
-
- Journal:
- Cambridge Quarterly of Healthcare Ethics / Volume 34 / Issue 2 / April 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 247-263
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Defect relation for holomorphic maps from complex discs into projective varieties and hypersurfaces
- Part of
-
- Journal:
- Proceedings of the Edinburgh Mathematical Society / Volume 68 / Issue 1 / February 2025
- Published online by Cambridge University Press:
- 22 November 2024, pp. 157-172
-
- Article
- Export citation
-
In this paper, we establish a second main theorem for holomorphic maps with finite growth index on complex discs intersecting arbitrary families of hypersurfaces (fixed and moving) in projective varieties, which gives an above bound of the sum of truncated defects. Our result also generalizes and improves many previous second main theorems for holomorphic maps from
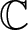 ${\mathbb{C}}$ intersecting hypersurfaces (moving and fixed) in projective varieties.
${\mathbb{C}}$ intersecting hypersurfaces (moving and fixed) in projective varieties.

Low-frequency unsteadiness in laminar separation bubbles
-
- Journal:
- Journal of Fluid Mechanics / Volume 999 / 25 November 2024
- Published online by Cambridge University Press:
- 22 November 2024, A99
-
- Article
- Export citation
-
Low-frequency phenomena in an incompressible pressure-induced laminar separation bubble (LSB) on a flat plate is investigated using direct numerical simulation. The LSB configuration of Spalart and Strelets (J. Fluid Mech., vol. 403, 2000, pp. 329–349) is used. Wall pressure spectra indicate low-frequency-flapping
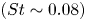 $(St \sim 0.08)$ and high-frequency-shedding
$(St \sim 0.08)$ and high-frequency-shedding 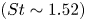 $(St \sim 1.52)$ regimes. Conditional velocity averages based on the fraction of reversed flow reveal the low frequency as an expansion/contraction of the LSB. While the high frequency only exhibits exponential growth within the LSB up to breakdown of the spanwise rollers, the low frequency and velocity fluctuations exhibit exponential growth upstream of separation. Instantaneous flow fields reveal large streamwise streaky structures forming within the LSB and extending past reattachment, much like high and low speed streaks in turbulent boundary layers. A predominance of sweep-like events (
$(St \sim 1.52)$ regimes. Conditional velocity averages based on the fraction of reversed flow reveal the low frequency as an expansion/contraction of the LSB. While the high frequency only exhibits exponential growth within the LSB up to breakdown of the spanwise rollers, the low frequency and velocity fluctuations exhibit exponential growth upstream of separation. Instantaneous flow fields reveal large streamwise streaky structures forming within the LSB and extending past reattachment, much like high and low speed streaks in turbulent boundary layers. A predominance of sweep-like events (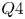 $Q4$) is observed during contraction and of ejection-like events (
$Q4$) is observed during contraction and of ejection-like events (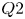 $Q2$) during expansion. These motions appear as dominant low-frequency modes in three-dimensional proper orthogonal and dynamic mode decompositions, exhibiting spatial amplification from separation to reattachment. The advection of a group of spanwise alternating streaky structures past the LSB results in an overall contraction after which the bubble expands to its ‘unforced’ state in the absence of the streaks. The low frequency then corresponds to the time it takes for streaks to form, amplify and advect past the LSB from separation to reattachment. This behaviour is linked to the mean flow deformation reported by Marxen and Rist (J. Fluid Mech., vol. 660, 2010, pp. 37–54), where the presence of streaks results in reduced mean bubble size. The formation of these streaky structures, in the absence of free stream turbulence, may be attributed to an absolute instability of the LSB due to the development of a secondary bubble within the primary.
$Q2$) during expansion. These motions appear as dominant low-frequency modes in three-dimensional proper orthogonal and dynamic mode decompositions, exhibiting spatial amplification from separation to reattachment. The advection of a group of spanwise alternating streaky structures past the LSB results in an overall contraction after which the bubble expands to its ‘unforced’ state in the absence of the streaks. The low frequency then corresponds to the time it takes for streaks to form, amplify and advect past the LSB from separation to reattachment. This behaviour is linked to the mean flow deformation reported by Marxen and Rist (J. Fluid Mech., vol. 660, 2010, pp. 37–54), where the presence of streaks results in reduced mean bubble size. The formation of these streaky structures, in the absence of free stream turbulence, may be attributed to an absolute instability of the LSB due to the development of a secondary bubble within the primary.