Refine search
Actions for selected content:
14 results
Host–pathogen interactions and genetic selection for resilience to post-weaning diarrhea in piglets
-
- Journal:
- Animal Nutriomics / Volume 3 / 2026
- Published online by Cambridge University Press:
- 09 February 2026, e16
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Modeling and analysis of nonlinear dynamics of axisymmetric vector nozzle based on deep neural network
-
- Journal:
- The Aeronautical Journal / Volume 129 / Issue 1335 / May 2025
- Published online by Cambridge University Press:
- 11 November 2024, pp. 1167-1181
-
- Article
- Export citation
EHMMQA: English, Hindi, and Marathi multilingual question answering framework using deep learning
-
- Journal:
- Natural Language Processing / Volume 31 / Issue 2 / March 2025
- Published online by Cambridge University Press:
- 24 May 2024, pp. 346-374
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Quantitative analysis of lateral root development with time-lapse imaging and deep neural network
-
- Journal:
- Quantitative Plant Biology / Volume 5 / 2024
- Published online by Cambridge University Press:
- 13 February 2024, e1
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Rapid generation of time-optimal rendezvous trajectory based on convex optimisation and DNN
-
- Journal:
- The Aeronautical Journal / Volume 128 / Issue 1324 / June 2024
- Published online by Cambridge University Press:
- 09 January 2024, pp. 1262-1283
-
- Article
- Export citation
2 - Deep Learning Background
-
- Book:
- Adversarial Learning and Secure AI
- Published online:
- 07 September 2023
- Print publication:
- 31 August 2023, pp 19-55
-
- Chapter
- Export citation
Describe the house and I will tell you the price: House price prediction with textual description data
-
- Journal:
- Natural Language Engineering / Volume 30 / Issue 4 / July 2024
- Published online by Cambridge University Press:
- 18 July 2023, pp. 661-695
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
House price prediction is an important problem that could benefit home buyers and sellers. Traditional models for house price prediction use numerical attributes such as the number of rooms but disregard the house description text. The recent developments in text processing suggest these can be valuable attributes, which motivated us to use house descriptions. This paper focuses on the house asking/advertising price and studies the impact of using house description texts to predict the final house price. To achieve this, we collected a large and diverse set of attributes on house postings, including the house advertising price. Then, we compare the performance of three scenarios: using only the house description, only numeric attributes, or both. We processed the description text through three word embedding techniques: TF-IDF, Word2Vec, and BERT. Four regression algorithms are trained using only textual data, non-textual data, or both. Our results show that by using exclusively the description data with Word2Vec and a Deep Learning model, we can achieve good performance. However, the best overall performance is obtained when using both textual and non-textual features. An
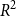 $R^2$ of 0.7904 is achieved by the deep learning model using only description data on the testing data. This clearly indicates that using the house description text alone is a strong predictor for the house price. However, when observing the RMSE on the test data, the best model was gradient boosting using both numeric and description data. Overall, we observe that combining the textual and non-textual features improves the learned model and provides performance benefits when compared against using only one of the feature types. We also provide a freely available application for house price prediction, which is solely based on a house text description and uses our final developed model with Word2Vec and Deep Learning to predict the house price.
$R^2$ of 0.7904 is achieved by the deep learning model using only description data on the testing data. This clearly indicates that using the house description text alone is a strong predictor for the house price. However, when observing the RMSE on the test data, the best model was gradient boosting using both numeric and description data. Overall, we observe that combining the textual and non-textual features improves the learned model and provides performance benefits when compared against using only one of the feature types. We also provide a freely available application for house price prediction, which is solely based on a house text description and uses our final developed model with Word2Vec and Deep Learning to predict the house price.
9 - Deep Learning
- from Part II - Cognitive Modeling Paradigms
-
-
- Book:
- The Cambridge Handbook of Computational Cognitive Sciences
- Published online:
- 21 April 2023
- Print publication:
- 11 May 2023, pp 301-349
-
- Chapter
- Export citation
15 - Deep Learning
-
- Book:
- Introduction to Environmental Data Science
- Published online:
- 23 March 2023
- Print publication:
- 23 March 2023, pp 494-517
-
- Chapter
- Export citation
A deep reinforcement learning-based approach to onboard trajectory generation for hypersonic vehicles
-
- Journal:
- The Aeronautical Journal / Volume 127 / Issue 1315 / September 2023
- Published online by Cambridge University Press:
- 08 February 2023, pp. 1638-1658
-
- Article
- Export citation
Effective multi-dialectal arabic POS tagging
-
- Journal:
- Natural Language Engineering / Volume 26 / Issue 6 / November 2020
- Published online by Cambridge University Press:
- 14 April 2020, pp. 677-690
-
- Article
- Export citation
Daily activity recognition based on recurrent neural network using multi-modal signals
- Part of
-
- Journal:
- APSIPA Transactions on Signal and Information Processing / Volume 7 / 2018
- Published online by Cambridge University Press:
- 01 January 2018, e21
- Print publication:
- 2018
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
