Refine search
Actions for selected content:
106116 results in Materials Science
Advances in Material Science and Applications
- Bulk (Silicon) vs Nano (Graphene)
- Coming soon
-
- Expected online publication date:
- April 2026
- Print publication:
- 01 September 2027
-
- Book
- Export citation
Advances in Material Science and Applications
- Bulk (Silicon) vs Nano (Graphene)
- Coming soon
-
- Expected online publication date:
- March 2026
- Print publication:
- 01 September 2027
-
- Book
- Export citation
Calendar of Forthcoming Meetings
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 26 December 2025, p. 419
-
- Article
-
- You have access
- HTML
- Export citation
PDJ volume 40 issue 4 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 26 December 2025, pp. f1-f4
-
- Article
-
- You have access
- Export citation
Our last issue with Cambridge University Press
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 26 December 2025, p. 256
-
- Article
-
- You have access
- HTML
- Export citation
Calendar of Short Courses and Workshops
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 26 December 2025, p. 420
-
- Article
-
- You have access
- HTML
- Export citation
PDJ volume 40 issue 4 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 26 December 2025, pp. b1-b2
-
- Article
-
- You have access
- Export citation
Crystal structure of valganciclovir hydrochloride, C14H23N6O5Cl
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 26 December 2025, pp. 304-312
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Effect of high-energy mixing on hydration of amorphous and crystalline Portland cement phases: an X-ray diffraction study
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 26 December 2025, pp. 279-284
-
- Article
- Export citation
Tetragonal–cubic phase transition in RbGaSi2O6 synthetic leucite analogue
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 05 December 2025, pp. 294-303
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The leucite group structures are tetrahedrally coordinated silicate framework structures with some of the silicate framework cations partially replaced by divalent or trivalent cations. These structures have general formulae A2BSi5O12 and ACSi2O6, where A is a monovalent alkali metal cation, B is a divalent cation, and C is a trivalent cation. These leucites can have crystal structures in several different space groups, dependent on stoichiometry, synthesis conditions, and temperature. Phase transitions are known for temperature changes. This paper reports a high-temperature X-ray powder diffraction study on RbGaSi2O6, which shows a phase transition from I41/a tetragonal to Ia
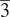 d cubic on heating from room temperature to 733 K. On cooling to room temperature, the crystal structure reverts to I41/a tetragonal.
d cubic on heating from room temperature to 733 K. On cooling to room temperature, the crystal structure reverts to I41/a tetragonal.
 d
dThe crystal structures of methoxmetamine hydrochloride and methoxetamine hydrochloride determined from laboratory X-ray powder diffraction data contained in the Powder Diffraction File™
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 01 December 2025, pp. 257-268
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
X-ray diffraction pattern analysis of CrFeCoNi high-entropy alloy deposited via cold spray
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 28 November 2025, pp. 269-278
-
- Article
- Export citation
Crystal structure of resmetirom heminonahydrate Form CSI, C17H12Cl2N6O4(H2O)4
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 28 November 2025, pp. 285-293
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure of fluvoxamine maleate, (C15H22F3N2O2)(HC4H2O4)
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 27 November 2025, pp. 313-320
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Proposed crystal structure of dequalinium chloride Form A, C30H40N4Cl2
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 21 November 2025, pp. 328-334
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Proposed crystal structure of protriptyline hydrochloride Form A, C19H22NCl
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 21 November 2025, pp. 335-341
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure of racemic afoxolaner, C26H17ClF9N3O3
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 18 November 2025, pp. 321-327
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
74th Annual Denver X-ray Conference Report
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 18 November 2025, pp. 414-418
-
- Article
- Export citation
Calendar of Forthcoming Meetings
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 3 / September 2025
- Published online by Cambridge University Press:
- 13 November 2025, pp. 253-254
-
- Article
-
- You have access
- HTML
- Export citation
The “chymistry” of Isaac Newton: a new Cu-acetate compound formed via crystallization
-
- Journal:
- Powder Diffraction / Volume 40 / Issue 3 / September 2025
- Published online by Cambridge University Press:
- 13 November 2025, pp. 183-189
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
