Refine search
Actions for selected content:
106117 results in Materials Science
Appendix D: - Analytical model for SMA fiber/Al matrix composite
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 478-482
-
- Chapter
- Export citation
6 - Bio-inspired designs of sensors, actuators
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 369-435
-
- Chapter
- Export citation
JMR volume 31 issue 19 Cover and Back matter
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 19 / 14 October 2016
- Published online by Cambridge University Press:
- 13 October 2016, pp. b1-b3
- Print publication:
- 14 October 2016
-
- Article
-
- You have access
- Export citation
Subject Index
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 519-524
-
- Chapter
- Export citation
References
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 486-511
-
- Chapter
- Export citation
Appendix C: - Polymer gel modeling, calculation of potentials in regions I-III in Nafion-based IPMC
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 474-477
-
- Chapter
- Export citation
7 - Design of autonomous systems
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 436-458
-
- Chapter
- Export citation
Preface
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp ix-xi
-
- Chapter
- Export citation
5 - Synthetic active materials and actuators
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 255-368
-
- Chapter
- Export citation
Appendix A: - Classical lamination theory under hygroscopic strains
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 459-469
-
- Chapter
- Export citation
4 - Synthetic sensing materials and sensors
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 208-254
-
- Chapter
- Export citation
3 - Sensory and motor systems of the living world
-
- Book:
- Bioinspired Actuators and Sensors
- Published online:
- 20 October 2016
- Print publication:
- 13 October 2016, pp 44-207
-
- Chapter
- Export citation
Crystal chemistry and X-ray diffraction patterns for Co(Nix Zn1−x )Nb4O12 (x = 0.2, 0.4, 0.6, 0.8)
-
- Journal:
- Powder Diffraction / Volume 31 / Issue 4 / December 2016
- Published online by Cambridge University Press:
- 12 October 2016, pp. 279-284
-
- Article
- Export citation
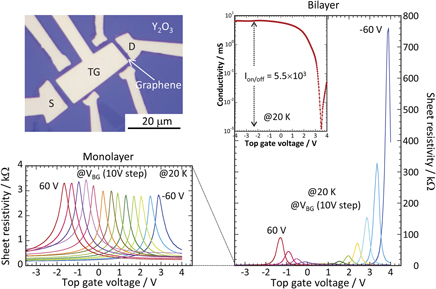
Graphene field-effect transistor application-electric band structure of graphene in transistor structure extracted from quantum capacitance
-
- Journal:
- Journal of Materials Research / Volume 32 / Issue 1 / 13 January 2017
- Published online by Cambridge University Press:
- 11 October 2016, pp. 64-72
- Print publication:
- 13 January 2017
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Analysis of hot deformation behavior and microstructure evolution of as-cast SiC nanoparticles reinforced magnesium matrix composite
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 21 / 14 November 2016
- Published online by Cambridge University Press:
- 11 October 2016, pp. 3437-3447
- Print publication:
- 14 November 2016
-
- Article
- Export citation

Microstructure and mechanical properties of Mo2Ni3Si–Al2O3 nanocomposite synthesized by mechanical alloying
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 21 / 14 November 2016
- Published online by Cambridge University Press:
- 11 October 2016, pp. 3352-3359
- Print publication:
- 14 November 2016
-
- Article
- Export citation

Development of bioceramic bone scaffolds by introducing triple liquid phases
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 22 / 28 November 2016
- Published online by Cambridge University Press:
- 11 October 2016, pp. 3498-3505
- Print publication:
- 28 November 2016
-
- Article
- Export citation

Research on the oxidation behavior of novel γ/γ′-strengthened Co–9Al–10W alloys combined with chromium and rare earth elements
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 21 / 14 November 2016
- Published online by Cambridge University Press:
- 11 October 2016, pp. 3332-3344
- Print publication:
- 14 November 2016
-
- Article
- Export citation
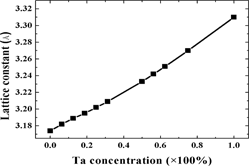
The effect of tantalum (Ta) doping on mechanical properties of tungsten (W): A first-principles study
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 21 / 14 November 2016
- Published online by Cambridge University Press:
- 11 October 2016, pp. 3401-3408
- Print publication:
- 14 November 2016
-
- Article
- Export citation

An effect of crystal tilt on the determination of ions displacements in perovskite oxides under BF/HAADF-STEM imaging mode
-
- Journal:
- Journal of Materials Research / Volume 32 / Issue 5 / 14 March 2017
- Published online by Cambridge University Press:
- 10 October 2016, pp. 947-956
- Print publication:
- 14 March 2017
-
- Article
- Export citation
