Refine listing
Actions for selected content:
1418918 results in Open Access
Reburials of Eminent Masters: The Construction of Quanzhen Daoist Lineages in North China under Mongol Rule
-
- Journal:
- Journal of Chinese History / Volume 8 / Issue 2 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 297-326
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Rising and settling 2-D cylinders with centre-of-mass offset
-
- Journal:
- Journal of Fluid Mechanics / Volume 981 / 25 February 2024
- Published online by Cambridge University Press:
- 16 February 2024, A7
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The Tang legacy on the Silk Road during the Uighur era: urbanisation in the eastern Tianshan region during the ninth to thirteenth centuries
-
- Journal:
- Journal of the Royal Asiatic Society / Volume 34 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 377-398
- Print publication:
- April 2024
-
- Article
- Export citation
Discursive scaling of solidarity through difference: Experiences of African women in the African diaspora
-
- Journal:
- Language in Society / Volume 54 / Issue 3 / June 2025
- Published online by Cambridge University Press:
- 16 February 2024, pp. 491-513
- Print publication:
- June 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Trilobites from the Al Rose Formation (Lower Ordovician, Inyo Mountains, California)—faunas marginal to the Great Basin
-
- Journal:
- Journal of Paleontology / Volume 98 / Issue 4 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 644-657
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
A single-institutional experience with 36 children less than 5 kilograms supported with the Berlin Heart: Comparison of congenital versus acquired heart disease
-
- Journal:
- Cardiology in the Young / Volume 34 / Issue 6 / June 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 1342-1349
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Effects of the COVID-19 Pandemic on Submissions to Politics & Gender
-
- Journal:
- PS: Political Science & Politics / Volume 57 / Issue 3 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 420-426
- Print publication:
- July 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
By Any Means Necessary? How Black and White Americans Evaluate Protest Tactics in Response to a Police Killing
-
- Journal:
- Journal of Race, Ethnicity and Politics / Volume 9 / Issue 2 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 235-258
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Holocene relative sea-level changes along the Caribbean and Pacific coasts of northwestern South America
-
- Journal:
- Quaternary Research / Volume 119 / May 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 28-43
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
BJN volume 131 issue 6 Cover and Front matter
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 6 / 28 March 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. f1-f2
- Print publication:
- 28 March 2024
-
- Article
-
- You have access
- Export citation
Association between dietary macronutrient composition and plasma one-carbon metabolites and B-vitamin cofactors in patients with stable angina pectoris
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 10 / 28 May 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 1678-1690
- Print publication:
- 28 May 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Rights and Wrongs in Talk of Mind-Reading Technology
-
- Journal:
- Cambridge Quarterly of Healthcare Ethics / Volume 33 / Issue 4 / October 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 521-531
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Introduction: Pandemic and Post-Pandemic Publication Patterns in Political Science
-
- Journal:
- PS: Political Science & Politics / Volume 57 / Issue 3 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 403-407
- Print publication:
- July 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Applied Probability Trust prizes 2023
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 16 February 2024, p. 368
- Print publication:
- March 2024
-
- Article
- Export citation
On a conjecture of Conlon, Fox, and Wigderson
- Part of
-
- Journal:
- Combinatorics, Probability and Computing / Volume 33 / Issue 4 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 432-445
-
- Article
- Export citation
-
For graphs
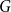 $G$ and
$G$ and 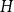 $H$, the Ramsey number
$H$, the Ramsey number 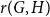 $r(G,H)$ is the smallest positive integer
$r(G,H)$ is the smallest positive integer 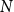 $N$ such that any red/blue edge colouring of the complete graph
$N$ such that any red/blue edge colouring of the complete graph 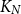 $K_N$ contains either a red
$K_N$ contains either a red 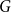 $G$ or a blue
$G$ or a blue 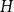 $H$. A book
$H$. A book 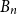 $B_n$ is a graph consisting of
$B_n$ is a graph consisting of  $n$ triangles all sharing a common edge.
$n$ triangles all sharing a common edge.Recently, Conlon, Fox, and Wigderson conjectured that for any
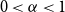 $0\lt \alpha \lt 1$, the random lower bound
$0\lt \alpha \lt 1$, the random lower bound 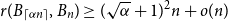 $r(B_{\lceil \alpha n\rceil },B_n)\ge (\sqrt{\alpha }+1)^2n+o(n)$ is not tight. In other words, there exists some constant
$r(B_{\lceil \alpha n\rceil },B_n)\ge (\sqrt{\alpha }+1)^2n+o(n)$ is not tight. In other words, there exists some constant 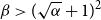 $\beta \gt (\sqrt{\alpha }+1)^2$ such that
$\beta \gt (\sqrt{\alpha }+1)^2$ such that 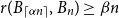 $r(B_{\lceil \alpha n\rceil },B_n)\ge \beta n$ for all sufficiently large
$r(B_{\lceil \alpha n\rceil },B_n)\ge \beta n$ for all sufficiently large  $n$. This conjecture holds for every
$n$. This conjecture holds for every 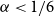 $\alpha \lt 1/6$ by a result of Nikiforov and Rousseau from 2005, which says that in this range
$\alpha \lt 1/6$ by a result of Nikiforov and Rousseau from 2005, which says that in this range 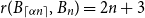 $r(B_{\lceil \alpha n\rceil },B_n)=2n+3$ for all sufficiently large
$r(B_{\lceil \alpha n\rceil },B_n)=2n+3$ for all sufficiently large  $n$.
$n$.We disprove the conjecture of Conlon, Fox, and Wigderson. Indeed, we show that the random lower bound is asymptotically tight for every
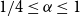 $1/4\leq \alpha \leq 1$. Moreover, we show that for any
$1/4\leq \alpha \leq 1$. Moreover, we show that for any 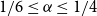 $1/6\leq \alpha \le 1/4$ and large
$1/6\leq \alpha \le 1/4$ and large  $n$,
$n$, 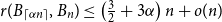 $r(B_{\lceil \alpha n\rceil }, B_n)\le \left (\frac 32+3\alpha \right ) n+o(n)$, where the inequality is asymptotically tight when
$r(B_{\lceil \alpha n\rceil }, B_n)\le \left (\frac 32+3\alpha \right ) n+o(n)$, where the inequality is asymptotically tight when 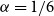 $\alpha =1/6$ or
$\alpha =1/6$ or 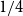 $1/4$. We also give a lower bound of
$1/4$. We also give a lower bound of 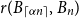 $r(B_{\lceil \alpha n\rceil }, B_n)$ for
$r(B_{\lceil \alpha n\rceil }, B_n)$ for 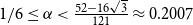 $1/6\le \alpha \lt \frac{52-16\sqrt{3}}{121}\approx 0.2007$, showing that the random lower bound is not tight, i.e., the conjecture of Conlon, Fox, and Wigderson holds in this interval.
$1/6\le \alpha \lt \frac{52-16\sqrt{3}}{121}\approx 0.2007$, showing that the random lower bound is not tight, i.e., the conjecture of Conlon, Fox, and Wigderson holds in this interval.
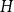
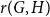
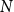
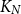
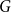
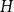
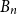

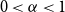
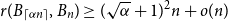
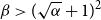
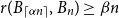

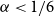
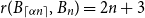

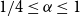
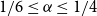

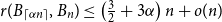
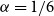
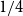
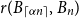
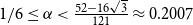
The restricted quantum double of the Yangian
- Part of
-
- Journal:
- Canadian Journal of Mathematics / Volume 77 / Issue 3 / June 2025
- Published online by Cambridge University Press:
- 16 February 2024, pp. 770-841
- Print publication:
- June 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Let
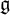 $\mathfrak {g}$ be a complex semisimple Lie algebra with associated Yangian
$\mathfrak {g}$ be a complex semisimple Lie algebra with associated Yangian 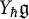 $Y_{\hbar }\mathfrak {g}$. In the mid-1990s, Khoroshkin and Tolstoy formulated a conjecture which asserts that the algebra
$Y_{\hbar }\mathfrak {g}$. In the mid-1990s, Khoroshkin and Tolstoy formulated a conjecture which asserts that the algebra 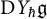 $\mathrm {D}Y_{\hbar }\mathfrak {g}$ obtained by doubling the generators of
$\mathrm {D}Y_{\hbar }\mathfrak {g}$ obtained by doubling the generators of 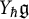 $Y_{\hbar }\mathfrak {g}$, called the Yangian double, provides a realization of the quantum double of the Yangian. We provide a uniform proof of this conjecture over
$Y_{\hbar }\mathfrak {g}$, called the Yangian double, provides a realization of the quantum double of the Yangian. We provide a uniform proof of this conjecture over 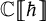 $\mathbb {C}[\kern-1.2pt\![{\hbar }]\!\kern-1.2pt]$ which is compatible with the theory of quantized enveloping algebras. As a by-product, we identify the universal R-matrix of the Yangian with the canonical element defined by the pairing between the Yangian and its restricted dual.
$\mathbb {C}[\kern-1.2pt\![{\hbar }]\!\kern-1.2pt]$ which is compatible with the theory of quantized enveloping algebras. As a by-product, we identify the universal R-matrix of the Yangian with the canonical element defined by the pairing between the Yangian and its restricted dual.
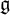
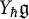
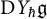
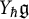
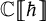
Optimizing Disaster Response: Structuring the Medical Branch Association’s Committees for Earthquake Emergencies
-
- Journal:
- Disaster Medicine and Public Health Preparedness / Volume 18 / 2024
- Published online by Cambridge University Press:
- 16 February 2024, e24
-
- Article
-
- You have access
- HTML
- Export citation
JPR volume 61 issue 1 Cover and Front matter
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. f1-f2
- Print publication:
- March 2024
-
- Article
-
- You have access
- Export citation
Calico Madams and South Sea Cheats: Global Trade, Finance, and Popular Protest in Early Hanoverian England
-
- Journal:
- Journal of British Studies / Volume 63 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 114-138
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
On twisted group ring isomorphism problem for p-groups
- Part of
-
- Journal:
- Glasgow Mathematical Journal / Volume 66 / Issue 2 / May 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 368-381
- Print publication:
- May 2024
-
- Article
- Export citation
-
In this article, we explore the problem of determining isomorphisms between the twisted complex group algebras of finite
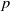 $p$-groups. This problem bears similarity to the classical group algebra isomorphism problem and has been recently examined by Margolis-Schnabel. Our focus lies on a specific invariant, referred to as the generalized corank, which relates to the twisted complex group algebra isomorphism problem. We provide a solution for non-abelian
$p$-groups. This problem bears similarity to the classical group algebra isomorphism problem and has been recently examined by Margolis-Schnabel. Our focus lies on a specific invariant, referred to as the generalized corank, which relates to the twisted complex group algebra isomorphism problem. We provide a solution for non-abelian 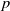 $p$-groups with generalized corank at most three.
$p$-groups with generalized corank at most three.