Refine listing
Actions for selected content:
1418153 results in Open Access
How to measure social health in the context of cognitive decline and dementia - A systematic review on instruments.
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 15-16
-
- Article
-
- You have access
- Export citation
Workshop 3: Develop, implement and evaluate technology for social health in dementia: lessons in best practice from the European DISTINCT network
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 2-3
-
- Article
-
- You have access
- Export citation
P136: Human Rights and Quality Standards for Services in Dementia Care
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 178-179
-
- Article
-
- You have access
- Export citation
Evaluating the Forward with Dementia Campaign in Five Countries
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 30-31
-
- Article
-
- You have access
- Export citation
Transgenderism, Othering and Third Way Buddhist Monasticism in Chiang Mai, Thailand
-
- Journal:
- TRaNS: Trans-Regional and -National Studies of Southeast Asia / Volume 12 / Issue 2 / November 2024
- Published online by Cambridge University Press:
- 02 February 2024, pp. 173-185
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Using Virtual Reality to Facilitate Reminiscence Therapy for People with Dementia
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 42
-
- Article
-
- You have access
- Export citation
P9: Clinical and Sociodemographic Factors Associated with Suicidal Risk in Older Adults in Latin America
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 109
-
- Article
-
- You have access
- Export citation
P132: Efficacy of Vortioxetine in Major Depression in Terminal Cancer. About a Case
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 177
-
- Article
-
- You have access
- Export citation
P151: A Chronic Grief Management-Intervention-Video: A Profile of the Sample
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 218
-
- Article
-
- You have access
- Export citation
P17: What happens if your colleague was the first person who notice that you have early-onset dementia?
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 236-237
-
- Article
-
- You have access
- Export citation
P120: Peer groups that support the mental health of older adults
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 213-214
-
- Article
-
- You have access
- Export citation
P121: Efficacy of adjunctive therapy of zonisamide versus increased dose of levodopa for motor symptom in DLB parkinsonism: a randomized, controlled, non-inferiority study, DUEL Study
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 173
-
- Article
-
- You have access
- Export citation
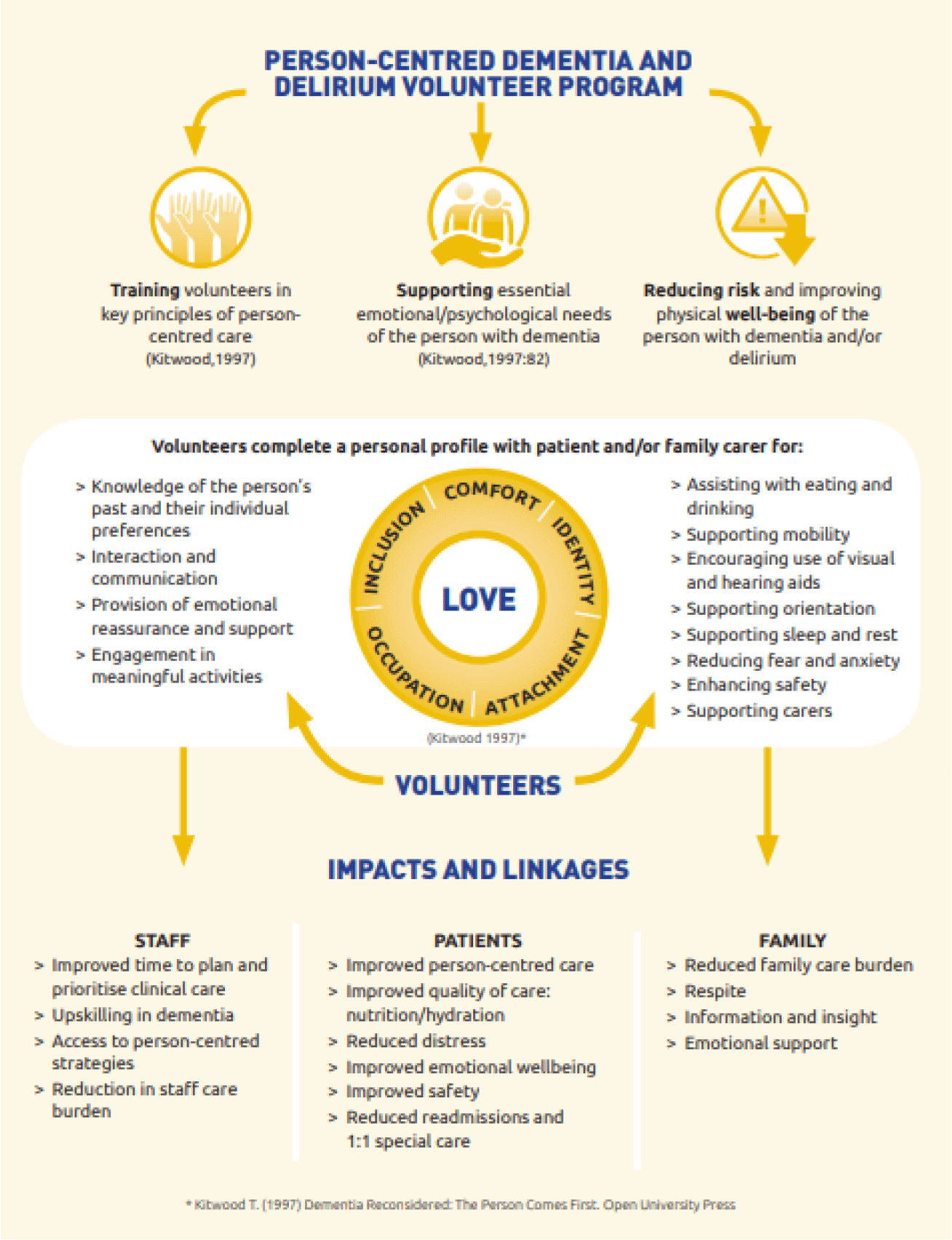
FC27: Golden Angels: The impact of volunteer support for patients with dementia and delirium in Australian rural hospitals
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 89-90
-
- Article
-
- You have access
- Export citation
-
Objectives:
evaluate the clinical outcomes for acute hospital patients with dementia, delirium or at risk for delirium supported by the person-centred volunteer program in australian rural hospitals.
Design:a non-randomised, mixed methods, controlled trial.
Participants:older adults admitted to 7 rural acute hospitals. Intervention (n=270) patients were >65 years with a diagnosis of dementia and/or delirium or had risk factors for delirium and received volunteer services. Family carers (n=83) of intervention patients were interviewed. Staff survey and focus groups. Control (n=188) patients were randomly drawn from patients admitted to the same hospital 12 months prior to the volunteer program who would have met program eligibility criteria.
Intervention:trained volunteers provided 1:1 person centred care with a focus on nutrition and hydration support, hearing and visual aids, activities, and orientation.
Measures:medical record audits provided data on volunteer visits, diagnoses, length of stay (los), behavioural incidents, readmission, specialling, mortality, admission to residential care, falls, pressure ulcers and medication use.
Results:across all sites there was a significant reduction in rates of 1:1 specialling (p=.011) and 28 day readmission (p=.006) for patients receiving the volunteer intervention. Los was significantly shorter for the control group (p=.001). All other patient outcomes were equivalent for the intervention and control group (p>.05). Volunteers integrated themselves into the care team providing person-centred care, increased safety and quality of care and were an “extra pair of hands”, reducing care burden for staff and importantly for families: “for me, knowing someone was there … i can't even tell you what a benefit that was”. 98% of staff rated the program as supportive of them in their care
Enablers were clear processes for screening, training and supporting volunteers. Key challenges included initial role delineation, staff/volunteer trust and sustainability.
Conclusion:appropriately trained and supported volunteers are cost effective and can improve the safety and quality of care for hospitalised patients with cognitive impairment in rural hospitals.

P117: The role of hyperarousal for understanding the association among sleep problems and emotional symptoms in family caregivers of people with dementia. A network analysis approach.
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 132
-
- Article
-
- You have access
- Export citation
P158: Efficacy of a self-help cognitive-behavioral therapy (GSH-CBT) guided by lay providers for generalized anxiety disorder (GAD) in older adults: preliminary results
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 220
-
- Article
-
- You have access
- Export citation
Linear transmission between malleus and stapes in cases with incus necrosis
-
- Journal:
- The Journal of Laryngology & Otology / Volume 138 / Issue 6 / June 2024
- Published online by Cambridge University Press:
- 02 February 2024, pp. 634-637
- Print publication:
- June 2024
-
- Article
- Export citation
Older adults and digital skills
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, p. 23
-
- Article
-
- You have access
- Export citation
P108: Relational aspects in dementia family caregiving: exploring caregivers ́ self-perceived caring style and its correlates in the caregiving stress and coping process
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 127-128
-
- Article
-
- You have access
- Export citation
P199: Cluster analysis dissecting cognitive deficits in older adults with major depressive disorder and the association between neurofilament light chain
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 188-189
-
- Article
-
- You have access
- Export citation
P147: Internet-delivered guided self-help Acceptance and Commitment Therapy for family carers of people with dementia (iACT4CARERS)
-
- Journal:
- International Psychogeriatrics / Volume 35 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 02 February 2024, pp. 254-255
-
- Article
-
- You have access
- Export citation
