Refine search
Actions for selected content:
106117 results in Materials Science
X-ray diffraction and density functional theory studies of R(Fe0.5Co0.5)O3 (R = Pr, Nd, Sm, Eu, Gd)
-
- Journal:
- Powder Diffraction / Volume 31 / Issue 4 / December 2016
- Published online by Cambridge University Press:
- 20 September 2016, pp. 259-266
-
- Article
- Export citation
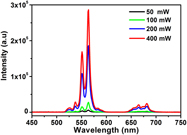
Electrical properties and upconversion luminescence of the Er3+-modified PZN–9PT crystals
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 19 / 14 October 2016
- Published online by Cambridge University Press:
- 20 September 2016, pp. 3044-3049
- Print publication:
- 14 October 2016
-
- Article
- Export citation
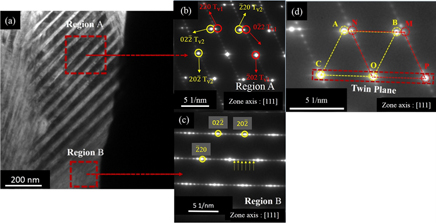
Structural transformations in highly oriented seven modulated martensite Ni–Mn–Ga thin films on an Al2O3
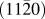 $\left( {11\bar 20} \right)$ substrate
$\left( {11\bar 20} \right)$ substrate
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 19 / 14 October 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 3016-3026
- Print publication:
- 14 October 2016
-
- Article
- Export citation
-
Highly oriented Ni–Mn–Ga thin film with multiple variants and room temperature orthorhombic martensite structure were prepared on a single crystalline Al2O3
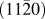 $\left( {11\bar 20} \right)$ substrate by DC magnetron sputtering. X-ray diffraction and rocking curve measurements reveal the film as (202)7M oriented with an excellent crystal quality (Δω = 1.8°). Spot-like pole figures indicate that the Ni–Mn–Ga film grows with a strong in-plane preferred orientation. An in-depth analysis of the measured pole figure reveals the presence of a retained austenite phase in the film. Two phase transformations, M S ∼345 K and T C ∼385 K, are observed and are attributed to first order structural transformation from cubic to orthorhombic, and second order phase transformation from ferromagnetic to paramagnetic, respectively. In situ high temperature x-ray diffraction measurements provide a clear indication of a thermally-induced martensite ↔ austenite reversible structural phase transformation in the film. The presence of martensite plates with seven modulated orthorhombic structure and adaptive nano-twins are some of the important microscopic features observed in the film with transmission electron microscopy investigations.
$\left( {11\bar 20} \right)$ substrate by DC magnetron sputtering. X-ray diffraction and rocking curve measurements reveal the film as (202)7M oriented with an excellent crystal quality (Δω = 1.8°). Spot-like pole figures indicate that the Ni–Mn–Ga film grows with a strong in-plane preferred orientation. An in-depth analysis of the measured pole figure reveals the presence of a retained austenite phase in the film. Two phase transformations, M S ∼345 K and T C ∼385 K, are observed and are attributed to first order structural transformation from cubic to orthorhombic, and second order phase transformation from ferromagnetic to paramagnetic, respectively. In situ high temperature x-ray diffraction measurements provide a clear indication of a thermally-induced martensite ↔ austenite reversible structural phase transformation in the film. The presence of martensite plates with seven modulated orthorhombic structure and adaptive nano-twins are some of the important microscopic features observed in the film with transmission electron microscopy investigations.
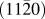
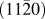

Study of activation energy and humidity sensing application of nanostructured Cu-doped ZnO thin films
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 20 / 28 October 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 3214-3222
- Print publication:
- 28 October 2016
-
- Article
- Export citation

Neutron reflectometry analysis of Li4Ti5O12/organic electrolyte interfaces: characterization of surface structure changes and lithium intercalation properties
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 20 / 28 October 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 3142-3150
- Print publication:
- 28 October 2016
-
- Article
- Export citation
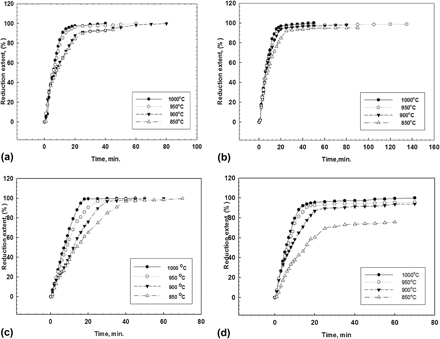
Synthesis of heavy tungsten alloys via powder reduction technique
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 19 / 14 October 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 2977-2986
- Print publication:
- 14 October 2016
-
- Article
- Export citation
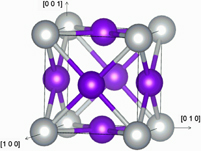
First-principles study of the structural, elastic and electronic properties of Pt3M alloys
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 19 / 14 October 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 2956-2963
- Print publication:
- 14 October 2016
-
- Article
- Export citation

Twin-mediated crystal growth
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 19 / 14 October 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 2936-2947
- Print publication:
- 14 October 2016
-
- Article
- Export citation
X-ray powder diffraction data for deferasirox, C21H15N3O4
-
- Journal:
- Powder Diffraction / Volume 31 / Issue 4 / December 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 298-300
-
- Article
- Export citation
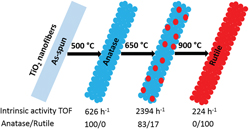
Enhanced intrinsic photocatalytic activity of TiO2 electrospun nanofibers based on temperature assisted manipulation of crystal phase ratios
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 19 / 14 October 2016
- Published online by Cambridge University Press:
- 19 September 2016, pp. 3036-3043
- Print publication:
- 14 October 2016
-
- Article
- Export citation
2 - Stress, strain, and isotropic elasticity
- from PART I - THEORETICAL BACKGROUND
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 21-47
-
- Chapter
- Export citation
8 - Dislocation geometry
- from PART III - DISLOCATIONS
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 209-243
-
- Chapter
- Export citation
Bibliography
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 507-514
-
- Chapter
- Export citation
13 - Grain boundary geometry
- from PART IV - GRAIN BOUNDARIES
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 433-454
-
- Chapter
- Export citation
3 - Statistical thermodynamics
- from PART I - THEORETICAL BACKGROUND
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 48-68
-
- Chapter
- Export citation
Frontmatter
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp i-iv
-
- Chapter
- Export citation
14 - Grain boundary mechanics
- from PART IV - GRAIN BOUNDARIES
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 455-487
-
- Chapter
- Export citation
4 - Point defect mechanics
- from PART II - POINT DEFECTS
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 71-100
-
- Chapter
- Export citation
12 - Dislocation core structure
- from PART III - DISLOCATIONS
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 395-430
-
- Chapter
- Export citation
5 - Point defect thermodynamics
- from PART II - POINT DEFECTS
-
- Book:
- Imperfections in Crystalline Solids
- Published online:
- 28 May 2018
- Print publication:
- 15 September 2016, pp 101-139
-
- Chapter
- Export citation
