Refine search
Actions for selected content:
106117 results in Materials Science
6 - A short historical interlude: the search for robust DNA motifs
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 88-96
-
- Chapter
- Export citation
10 - Combining structure and motion
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 172-185
-
- Chapter
- Export citation
2 - The design of DNA sequences for branched systems
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 11-27
-
- Chapter
- Export citation
5 - Experimental techniques
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 64-87
-
- Chapter
- Export citation
Preface
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp ix-x
-
- Chapter
- Export citation
13 - Not just plain vanilla DNA nanotechnology: other pairings, other backbones
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 213-230
-
- Chapter
- Export citation
11 - Self-replicating systems
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 186-197
-
- Chapter
- Export citation
12 - Computing with DNA
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 198-212
-
- Chapter
- Export citation

Ab initio investigation of the substitution effects of 2p elements on the electronic structure of γ-Fe4X (X = B, C, N, and O) in the ground state
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 07 January 2016, pp. 202-212
- Print publication:
- 28 January 2016
-
- Article
- Export citation
9 - DNA origami and DNA bricks
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 150-171
-
- Chapter
- Export citation
Index
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 251-257
-
- Chapter
- Export citation
Contents
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp vii-viii
-
- Chapter
- Export citation
Frontmatter
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp i-vi
-
- Chapter
- Export citation

The stabilities and electronic structures of Aln Si12−n N12 (n = 0, 1, 2, and 4)
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 07 January 2016, pp. 241-249
- Print publication:
- 28 January 2016
-
- Article
- Export citation
8 - DNA nanomechanical devices
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 130-149
-
- Chapter
- Export citation
4 - Single-stranded DNA topology and motif design
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 44-63
-
- Chapter
- Export citation

Measurement of local mechanical properties using multiple indentations by a special conical indenter and error analysis
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 06 January 2016, pp. 259-273
- Print publication:
- 28 January 2016
-
- Article
- Export citation

Magnesium nanocomposite: Effect of melt dispersion of different oxides nano particles
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 06 January 2016, pp. 100-108
- Print publication:
- 14 January 2016
-
- Article
- Export citation
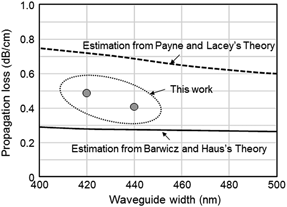
Low-loss silicon wire waveguides for optical integrated circuits
-
- Journal:
- MRS Communications / Volume 6 / Issue 1 / March 2016
- Published online by Cambridge University Press:
- 06 January 2016, pp. 9-15
- Print publication:
- March 2016
-
- Article
- Export citation

Measurement and analysis of friction and wear on electrodeposited coatings against a high carbon chrome steel ball
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 13 / 14 July 2016
- Published online by Cambridge University Press:
- 06 January 2016, pp. 1865-1872
- Print publication:
- 14 July 2016
-
- Article
- Export citation
