Refine listing
Actions for selected content:
Most read
This page lists the top ten most read articles for this journal based on the number of full text views and downloads recorded on Cambridge Core over the last 90 days. This list is updated on a daily basis.
Contents
De novo protein design, a retrospective
-
- Published online by Cambridge University Press:
- 11 February 2020, e3
-
- Article
-
- You have access
- HTML
- Export citation
Development of CRISPR-Cas systems for genome editing and beyond
-
- Published online by Cambridge University Press:
- 13 June 2019, e6
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Using multiscale molecular dynamics simulations to explore the fusion machinery underlying neurotransmitter release
-
- Published online by Cambridge University Press:
- 27 June 2025, e14
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
In silico ADME/T modelling for rational drug design
-
- Published online by Cambridge University Press:
- 02 September 2015, pp. 488-515
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
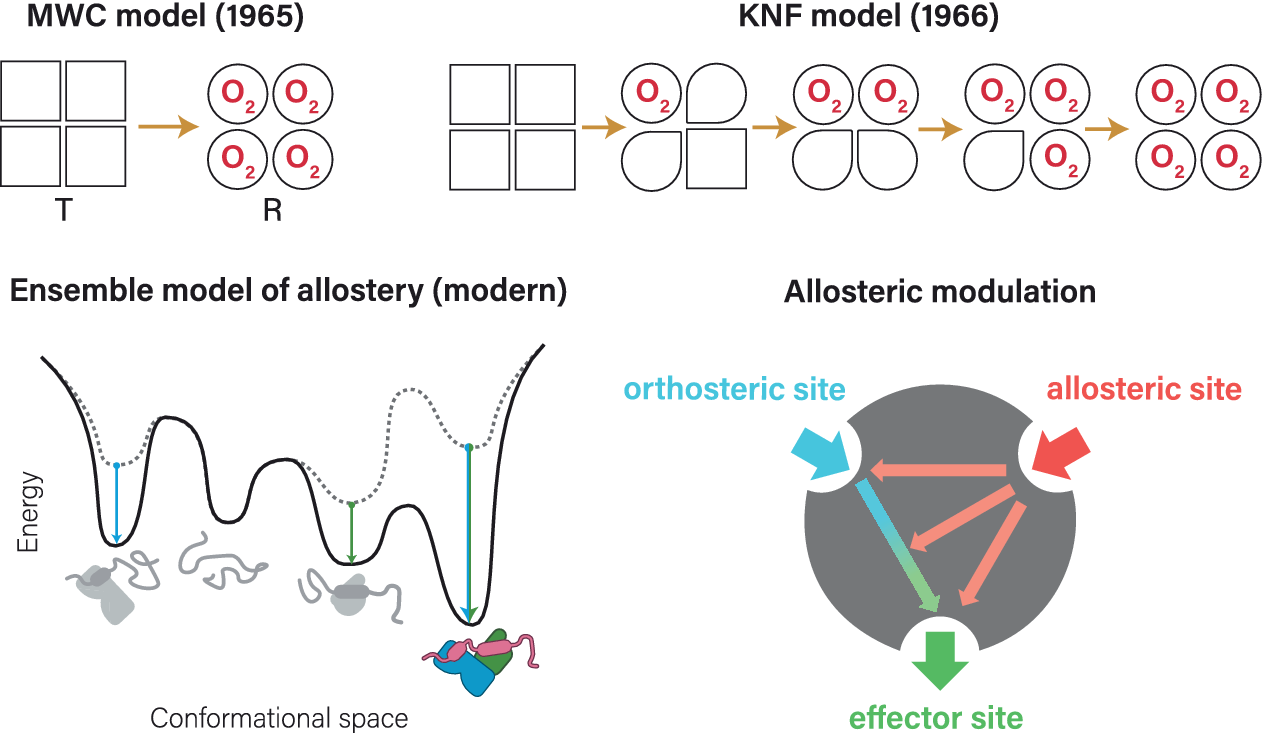
Allostery
-
- Published online by Cambridge University Press:
- 24 January 2025, e5
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Computational methods for binding site prediction on macromolecules
-
- Published online by Cambridge University Press:
- 12 March 2025, e12
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Hydrophobic interactions control the self-assembly of DNA and cellulose
-
- Published online by Cambridge University Press:
- 05 February 2021, e3
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Graph theory approaches for molecular dynamics simulations
-
- Published online by Cambridge University Press:
- 10 December 2024, e15
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Macromolecular room temperature crystallography
-
- Published online by Cambridge University Press:
- 08 January 2021, e1
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Acceleration of reaction in charged microdroplets
-
- Published online by Cambridge University Press:
- 16 July 2015, pp. 437-444
-
- Article
-
- You have access
- Open access
- HTML
- Export citation