Refine listing
Actions for selected content:
1419635 results in Open Access
INDEX
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 573-605
- Print publication:
- December 2024
-
- Article
- Export citation
APPENDIX 2 INVENTORY TAKEN AT BROOKE HOUSE, HOLBORN, 28 JULY 1643
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 509-519
- Print publication:
- December 2024
-
- Article
- Export citation
MAPS
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. ix-xi
- Print publication:
- December 2024
-
- Article
- Export citation
Is Intimate Partner Violence a Risk Factor for Alzheimer’s Disease?
-
- Journal:
- Canadian Journal of Neurological Sciences / Volume 52 / Issue 5 / September 2025
- Published online by Cambridge University Press:
- 26 November 2024, pp. 737-738
-
- Article
- Export citation
Optimal mixing via tensorization for random independent sets on arbitrary trees
- Part of
-
- Journal:
- Combinatorics, Probability and Computing / Volume 34 / Issue 2 / March 2025
- Published online by Cambridge University Press:
- 26 November 2024, pp. 259-275
-
- Article
- Export citation
-
We study the mixing time of the single-site update Markov chain, known as the Glauber dynamics, for generating a random independent set of a tree. Our focus is obtaining optimal convergence results for arbitrary trees. We consider the more general problem of sampling from the Gibbs distribution in the hard-core model where independent sets are weighted by a parameter
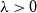 $\lambda \gt 0$; the special case
$\lambda \gt 0$; the special case 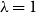 $\lambda =1$ corresponds to the uniform distribution over all independent sets. Previous work of Martinelli, Sinclair and Weitz (2004) obtained optimal mixing time bounds for the complete
$\lambda =1$ corresponds to the uniform distribution over all independent sets. Previous work of Martinelli, Sinclair and Weitz (2004) obtained optimal mixing time bounds for the complete 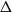 $\Delta$-regular tree for all
$\Delta$-regular tree for all 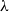 $\lambda$. However, Restrepo, Stefankovic, Vera, Vigoda, and Yang (2014) showed that for sufficiently large
$\lambda$. However, Restrepo, Stefankovic, Vera, Vigoda, and Yang (2014) showed that for sufficiently large 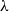 $\lambda$ there are bounded-degree trees where optimal mixing does not hold. Recent work of Eppstein and Frishberg (2022) proved a polynomial mixing time bound for the Glauber dynamics for arbitrary trees, and more generally for graphs of bounded tree-width.
$\lambda$ there are bounded-degree trees where optimal mixing does not hold. Recent work of Eppstein and Frishberg (2022) proved a polynomial mixing time bound for the Glauber dynamics for arbitrary trees, and more generally for graphs of bounded tree-width.We establish an optimal bound on the relaxation time (i.e., inverse spectral gap) of
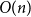 $O(n)$ for the Glauber dynamics for unweighted independent sets on arbitrary trees. We stress that our results hold for arbitrary trees and there is no dependence on the maximum degree
$O(n)$ for the Glauber dynamics for unweighted independent sets on arbitrary trees. We stress that our results hold for arbitrary trees and there is no dependence on the maximum degree 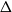 $\Delta$. Interestingly, our results extend (far) beyond the uniqueness threshold which is on the order
$\Delta$. Interestingly, our results extend (far) beyond the uniqueness threshold which is on the order 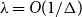 $\lambda =O(1/\Delta )$. Our proof approach is inspired by recent work on spectral independence. In fact, we prove that spectral independence holds with a constant independent of the maximum degree for any tree, but this does not imply mixing for general trees as the optimal mixing results of Chen, Liu, and Vigoda (2021) only apply for bounded-degree graphs. We instead utilize the combinatorial nature of independent sets to directly prove approximate tensorization of variance via a non-trivial inductive proof.
$\lambda =O(1/\Delta )$. Our proof approach is inspired by recent work on spectral independence. In fact, we prove that spectral independence holds with a constant independent of the maximum degree for any tree, but this does not imply mixing for general trees as the optimal mixing results of Chen, Liu, and Vigoda (2021) only apply for bounded-degree graphs. We instead utilize the combinatorial nature of independent sets to directly prove approximate tensorization of variance via a non-trivial inductive proof.
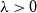
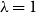
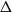
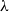
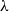
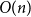
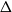
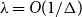
Generalized Ricci flow on aligned homogeneous spaces
- Part of
-
- Journal:
- Proceedings of the Edinburgh Mathematical Society / Volume 68 / Issue 1 / February 2025
- Published online by Cambridge University Press:
- 26 November 2024, pp. 226-246
-
- Article
- Export citation
-
The fixed points of the generalized Ricci flow are the Bismut Ricci flat (BRF) metrics, i.e., a generalized metric (g, H) on a manifold M, where g is a Riemannian metric and H a closed 3-form, such that H is g-harmonic and
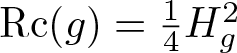 $\operatorname{Rc}(g)=\tfrac{1}{4} H_g^2$. Given two standard Einstein homogeneous spaces
$\operatorname{Rc}(g)=\tfrac{1}{4} H_g^2$. Given two standard Einstein homogeneous spaces 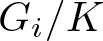 $G_i/K$, where each Gi is a compact simple Lie group and K is a closed subgroup of them holding some extra assumption, we consider
$G_i/K$, where each Gi is a compact simple Lie group and K is a closed subgroup of them holding some extra assumption, we consider  $M=G_1\times G_2/\Delta K$. Recently, Lauret and Will proved the existence of a BRF metric on any of these spaces. We proved that this metric is always asymptotically stable for the generalized Ricci flow on M among a subset of G-invariant metrics and, if
$M=G_1\times G_2/\Delta K$. Recently, Lauret and Will proved the existence of a BRF metric on any of these spaces. We proved that this metric is always asymptotically stable for the generalized Ricci flow on M among a subset of G-invariant metrics and, if  $G_1=G_2$, then it is globally stable.
$G_1=G_2$, then it is globally stable.
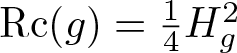
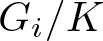


INTRODUCTION
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 1-32
- Print publication:
- December 2024
-
- Article
- Export citation
Widths of crossings in Poisson Boolean percolation
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 62 / Issue 2 / June 2025
- Published online by Cambridge University Press:
- 26 November 2024, pp. 603-627
- Print publication:
- June 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
We answer the following question: if the occupied (or vacant) set of a planar Poisson Boolean percolation model contains a crossing of an
 $n\times n$ square, how wide is this crossing? The answer depends on whether we consider the critical, sub-, or super-critical regime, and is different for the occupied and vacant sets.
$n\times n$ square, how wide is this crossing? The answer depends on whether we consider the critical, sub-, or super-critical regime, and is different for the occupied and vacant sets.

ACCOUNTS OF KATHERINE, LADY BROOKE, 1648–1649
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 447-502
- Print publication:
- December 2024
-
- Article
- Export citation

Quasi-steady transitions in confined convection
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 26 November 2024, A44
-
- Article
- Export citation
-
We study the effect of geometrical confinement on thermal convection by laboratory experiments and direct numerical simulations using Hele-Shaw geometries (typically the gap-to-height aspect ratio
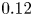 $0.12$) for the Prandtl number
$0.12$) for the Prandtl number 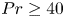 $Pr \geq 40$ and the Rayleigh number
$Pr \geq 40$ and the Rayleigh number 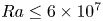 $Ra \leq 6 \times 10^7$. Under such strong unidirectional confinement, the convective flows are forced to squeeze within the narrow gap and exhibit unique spatiotemporal signatures, which contrast those in unconfined systems. With the increase of
$Ra \leq 6 \times 10^7$. Under such strong unidirectional confinement, the convective flows are forced to squeeze within the narrow gap and exhibit unique spatiotemporal signatures, which contrast those in unconfined systems. With the increase of 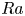 $Ra$, we identify that the system experiences five convective regimes that can be classified from two aspects, time dependency and flow dimensionality: (I) quasi-two-dimensional (quasi-2-D) steady flow; (II) quasi-2-D flow with oscillatory corner rolls; (III) three-dimensional (3-D) flow with oscillatory corner rolls; (IV) 3-D steady flow; and (V) 3-D time-dependent motion of plumes around sidewalls. Notably, unsteadiness does not emerge globally, but is localised near the sidewalls as oscillatory corner rolls, resulting in the regime transitions happening in a quasi-steady manner. We confirm that these regime transitions show less dependence on both
$Ra$, we identify that the system experiences five convective regimes that can be classified from two aspects, time dependency and flow dimensionality: (I) quasi-two-dimensional (quasi-2-D) steady flow; (II) quasi-2-D flow with oscillatory corner rolls; (III) three-dimensional (3-D) flow with oscillatory corner rolls; (IV) 3-D steady flow; and (V) 3-D time-dependent motion of plumes around sidewalls. Notably, unsteadiness does not emerge globally, but is localised near the sidewalls as oscillatory corner rolls, resulting in the regime transitions happening in a quasi-steady manner. We confirm that these regime transitions show less dependence on both 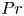 $Pr$ and the other (wider) horizontal scale of the geometry. Moreover, we find that a recently proposed criterion ‘degree of confinement’ (Noto et al., Proc. Natl Acad. Sci. USA, vol. 121, issue 28, 2024, e2403699121) successfully explains the emergence of 3-D structures, expanding its applicable range to smaller
$Pr$ and the other (wider) horizontal scale of the geometry. Moreover, we find that a recently proposed criterion ‘degree of confinement’ (Noto et al., Proc. Natl Acad. Sci. USA, vol. 121, issue 28, 2024, e2403699121) successfully explains the emergence of 3-D structures, expanding its applicable range to smaller 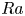 $Ra$. This study deepens the comprehension of the thermal convection emerging in tight geometries, impacting across disciplines, such as Earth and planetary science, and thermal engineering.
$Ra$. This study deepens the comprehension of the thermal convection emerging in tight geometries, impacting across disciplines, such as Earth and planetary science, and thermal engineering.
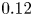
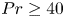
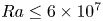
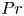
Beat the elite or concede defeat? Populist problem (re-)representations of international financial disputes
-
- Journal:
- Review of International Studies , First View
- Published online by Cambridge University Press:
- 26 November 2024, pp. 1-24
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
ACCOUNTS OF MAJOR JOSEPH HAWKSWORTH, 1641–1643
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 233-320
- Print publication:
- December 2024
-
- Article
- Export citation
ACCOUNTS OF KATHERINE, LADY BROOKE, 1644–1645
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 343-369
- Print publication:
- December 2024
-
- Article
- Export citation

Homogeneous turbophoresis of heavy inertial particles in turbulent flow
-
- Journal:
- Journal of Fluid Mechanics / Volume 999 / 25 November 2024
- Published online by Cambridge University Press:
- 26 November 2024, A83
-
- Article
- Export citation
-
Heavy particles suspended in turbulent flow possess inertia and are ejected from violent vortical structures by centrifugal forces. Once piled up along particle paths, this small-scale mechanism leads to an effective large-scale drift. This phenomenon, known as ‘turbophoresis’, causes particles to leave highly turbulent regions and migrate towards calmer regions, explaining why particles transported by non-homogeneous flows tend to concentrate near the minima of turbulent kinetic energy. It is demonstrated here that turbophoretic effects are just as crucial in statistically homogeneous flows. Although the average turbulent activity is uniform, instantaneous spatial fluctuations are responsible for inertial-range inhomogeneities in the particle distribution. Direct numerical simulations are used to probe particle accelerations, specifically how they correlate to local turbulent activity, yielding an effective coarse-grained dynamics that accounts for particle detachment from the fluid and ejection from excited regions through a space- and time-dependent non-Fickian diffusion. This leads to cast fluctuations in particle distributions in terms of a scale-dependent Péclet number
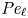 ${\textit {Pe}}_\ell$, which measures the importance of turbulent advection compared with inertial turbophoresis at a given scale
${\textit {Pe}}_\ell$, which measures the importance of turbulent advection compared with inertial turbophoresis at a given scale 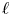 $\ell$. Multifractal statistics of energy dissipation indicate that
$\ell$. Multifractal statistics of energy dissipation indicate that 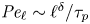 $ {\textit {Pe}}_\ell \sim \ell ^\delta /\tau _{p}$ with
$ {\textit {Pe}}_\ell \sim \ell ^\delta /\tau _{p}$ with 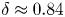 $\delta \approx 0.84$. Numerical simulations support this behaviour and emphasise the relevance of the turbophoretic Péclet number in characterising how particle distributions, including their radial distribution function, depends on
$\delta \approx 0.84$. Numerical simulations support this behaviour and emphasise the relevance of the turbophoretic Péclet number in characterising how particle distributions, including their radial distribution function, depends on 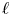 $\ell$. This approach also explains the presence of voids with inertial-range sizes, and the fact that their volumes have a non-trivial distribution with a power-law tail
$\ell$. This approach also explains the presence of voids with inertial-range sizes, and the fact that their volumes have a non-trivial distribution with a power-law tail 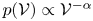 $p(\mathcal {V}) \propto \mathcal {V}^{-\alpha }$, with an exponent
$p(\mathcal {V}) \propto \mathcal {V}^{-\alpha }$, with an exponent 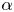 $\alpha$ that tends to 2 as
$\alpha$ that tends to 2 as 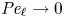 ${\textit {Pe}}_\ell \to 0$.
${\textit {Pe}}_\ell \to 0$.
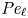
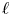
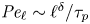
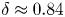
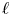
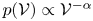
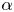
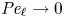
Charmaine Robson, Missionary Women, Leprosy and Indigenous Australians, 1936-1986 (Cham: Palgrave Macmillan, 2022), pp. VIII + 265, £79.50, eBook, ISBN: 978-3-031-05796-0
-
- Journal:
- Medical History / Volume 68 / Issue 4 / October 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 488-489
-
- Article
-
- You have access
- HTML
- Export citation
ACCOUNTS OF MAJOR JOSEPH HAWKSWORTH, 1640–1641
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, pp. 187-232
- Print publication:
- December 2024
-
- Article
- Export citation
ABBREVIATIONS
-
- Journal:
- Royal Historical Society Camden Fifth Series / Volume 68 / December 2024
- Published online by Cambridge University Press:
- 26 November 2024, p. xv
- Print publication:
- December 2024
-
- Article
- Export citation
The Epistemic Fata Morgana: Appropriation in the Institutional Context
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Lubrication-mediated rebounds off fluid baths
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 26 November 2024, R2
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
