Refine listing
Actions for selected content:
1416829 results in Open Access
Surface Structural Model for Ferrihydrite
-
- Journal:
- Clays and Clay Minerals / Volume 45 / Issue 3 / June 1997
- Published online by Cambridge University Press:
- 28 February 2024, pp. 448-460
-
- Article
- Export citation
Infrared Spectroscopic Analyses on the Nature of Water in Montmorillonite
-
- Journal:
- Clays and Clay Minerals / Volume 42 / Issue 6 / December 1994
- Published online by Cambridge University Press:
- 28 February 2024, pp. 702-716
-
- Article
- Export citation
The degree one Laguerre–Pólya class and the shuffle-word-embedding conjecture
- Part of
-
- Journal:
- Canadian Mathematical Bulletin / Volume 67 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 28 February 2024, pp. 760-767
- Print publication:
- September 2024
-
- Article
- Export citation
-
We discuss the class of functions, which are well approximated on compacta by the geometric mean of the eigenvalues of a unital (completely) positive map into a matrix algebra or more generally a type
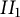 $II_1$ factor, using the notion of a Fuglede–Kadison determinant. In two variables, the two classes are the same, but in three or more noncommuting variables, there are generally functions arising from type
$II_1$ factor, using the notion of a Fuglede–Kadison determinant. In two variables, the two classes are the same, but in three or more noncommuting variables, there are generally functions arising from type 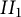 $II_1$ von Neumann algebras, due to the recently established failure of the Connes embedding conjecture. The question of whether or not approximability holds for scalar inputs is shown to be equivalent to a restricted form of the Connes embedding conjecture, the so-called shuffle-word-embedding conjecture.
$II_1$ von Neumann algebras, due to the recently established failure of the Connes embedding conjecture. The question of whether or not approximability holds for scalar inputs is shown to be equivalent to a restricted form of the Connes embedding conjecture, the so-called shuffle-word-embedding conjecture.
The Thermodynamic Status of Compositionally-Complex Clay Minerals: Discussion of “Clay Mineral Thermometry—A Critical Perspective”
-
- Journal:
- Clays and Clay Minerals / Volume 44 / Issue 4 / August 1996
- Published online by Cambridge University Press:
- 28 February 2024, pp. 560-568
-
- Article
- Export citation
Zeolite Distribution in Volcaniclastic Deep-sea Sediments from the Tonga Trench Margin (SW Pacific)
-
- Journal:
- Clays and Clay Minerals / Volume 43 / Issue 1 / February 1995
- Published online by Cambridge University Press:
- 28 February 2024, pp. 92-104
-
- Article
- Export citation
Determination of Mean Crystallite Dimensions from X-Ray Diffraction Peak Profiles: A Comparative Analysis of Synthetic Hematites
-
- Journal:
- Clays and Clay Minerals / Volume 47 / Issue 6 / December 1999
- Published online by Cambridge University Press:
- 28 February 2024, pp. 742-747
-
- Article
- Export citation
Clay Poultices in Salt Extraction from Ornamental Stones: A Statistical Approach
-
- Journal:
- Clays and Clay Minerals / Volume 49 / Issue 3 / June 2001
- Published online by Cambridge University Press:
- 28 February 2024, pp. 227-235
-
- Article
- Export citation
Kaolinite/Montmorillonite Resembles Beidellite
-
- Journal:
- Clays and Clay Minerals / Volume 42 / Issue 5 / October 1994
- Published online by Cambridge University Press:
- 28 February 2024, pp. 643-651
-
- Article
- Export citation
Adsorption of Nitrilotriacetate (NTA), Co and CoNTA By Gibbsite
-
- Journal:
- Clays and Clay Minerals / Volume 44 / Issue 6 / December 1996
- Published online by Cambridge University Press:
- 28 February 2024, pp. 757-768
-
- Article
- Export citation
The Kinetics of the Smectite to Illite Transformation in Cretaceous Bentonites, Cerro Negro, New Mexico
-
- Journal:
- Clays and Clay Minerals / Volume 47 / Issue 3 / June 1999
- Published online by Cambridge University Press:
- 28 February 2024, pp. 286-296
-
- Article
- Export citation
Mineral Metastability in the System Al2O3-SiO2-H2O: A Reply
-
- Journal:
- Clays and Clay Minerals / Volume 42 / Issue 1 / February 1994
- Published online by Cambridge University Press:
- 28 February 2024, pp. 102-105
-
- Article
- Export citation
Stirring Effects on Properties of Al Goethite Formed From Ferrihydrite
-
- Journal:
- Clays and Clay Minerals / Volume 46 / Issue 3 / June 1998
- Published online by Cambridge University Press:
- 28 February 2024, pp. 317-321
-
- Article
- Export citation
Role of Tartaric Acid in the Inhibition of the Formation of Al13 Tridecamer using Sulfate Precipitation
-
- Journal:
- Clays and Clay Minerals / Volume 47 / Issue 5 / October 1999
- Published online by Cambridge University Press:
- 28 February 2024, pp. 658-663
-
- Article
- Export citation
Stable Isotope Evolution of Volcanic Ash Layers During Diagenesis of the Miocene Monterey Formation, California
-
- Journal:
- Clays and Clay Minerals / Volume 47 / Issue 1 / February 1999
- Published online by Cambridge University Press:
- 28 February 2024, pp. 84-95
-
- Article
- Export citation
Molecular Structure Effects on Diffusion of Cations in Clays
-
- Journal:
- Clays and Clay Minerals / Volume 44 / Issue 3 / June 1996
- Published online by Cambridge University Press:
- 28 February 2024, pp. 370-380
-
- Article
- Export citation
Preferred Orientation of Phyllosilicates in Gulf Coast Mudstones and Relation to the Smectite-Illite Transition
-
- Journal:
- Clays and Clay Minerals / Volume 47 / Issue 4 / August 1999
- Published online by Cambridge University Press:
- 28 February 2024, pp. 495-504
-
- Article
- Export citation
Cooling Rate Dependency of the Formation of Smectite Crystals from a High-Pressure and High-Temperature Hydrous Melt
-
- Journal:
- Clays and Clay Minerals / Volume 43 / Issue 6 / December 1995
- Published online by Cambridge University Press:
- 28 February 2024, pp. 693-696
-
- Article
- Export citation
Nature of Structural Disorder in Natural Kaolinites: A New Model Based on Computer Simulation of Powder Diffraction Data and Electrostatic Energy Calculation
-
- Journal:
- Clays and Clay Minerals / Volume 43 / Issue 4 / August 1995
- Published online by Cambridge University Press:
- 28 February 2024, pp. 438-445
-
- Article
- Export citation
Bleaching Properties of Alumina-Pillared Acid-Activated Montmorillonite
-
- Journal:
- Clays and Clay Minerals / Volume 48 / Issue 5 / October 2000
- Published online by Cambridge University Press:
- 28 February 2024, pp. 549-556
-
- Article
- Export citation
Baseline Studies of the Clay Minerals Society Source Clays: Chemical Analysis by Inductively Coupled Plasma-Mass Spectroscopy (ICP-MS)
-
- Journal:
- Clays and Clay Minerals / Volume 49 / Issue 5 / October 2001
- Published online by Cambridge University Press:
- 28 February 2024, pp. 387-392
-
- Article
- Export citation
