Refine search
Actions for selected content:
106116 results in Materials Science
Reviews
-
- Book:
- Nucleation of Particles from the Gas Phase
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024, pp ii-ii
-
- Chapter
- Export citation
Symbols
-
- Book:
- Nucleation of Particles from the Gas Phase
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024, pp xiii-xviii
-
- Chapter
- Export citation
Preface
-
- Book:
- Nucleation of Particles from the Gas Phase
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024, pp xi-xii
-
- Chapter
- Export citation
2 - Single-Component Homogeneous Nucleation from a Supersaturated Vapor
-
- Book:
- Nucleation of Particles from the Gas Phase
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024, pp 14-31
-
- Chapter
- Export citation
1 - Introduction
-
- Book:
- Nucleation of Particles from the Gas Phase
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024, pp 1-13
-
- Chapter
- Export citation
3 - Classical Nucleation Theory
-
- Book:
- Nucleation of Particles from the Gas Phase
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024, pp 32-93
-
- Chapter
- Export citation
Copyright page
-
- Book:
- Nucleation of Particles from the Gas Phase
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024, pp iv-iv
-
- Chapter
- Export citation
Bimodal microstructural characterization of Si powder using X-ray diffraction: the role of peak shape
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 04 June 2024, pp. 119-131
-
- Article
- Export citation
Crystal chemistry and ionic conductivity of garnet-type solid-state electrolyte, Li5-xLa3(NbTa)O12-y
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 03 June 2024, pp. 191-205
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Nucleation of Particles from the Gas Phase
-
- Published online:
- 30 May 2024
- Print publication:
- 06 June 2024
Powder X-ray diffraction of acalabrutinib dihydrate Form III, C26H23N7O2(H2O)2
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 27 May 2024, pp. 283-286
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure of ribociclib hydrogen succinate, (C23H31N8O)(HC4H4O4)
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 27 May 2024, pp. 227-234
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
An introduction to linkage fabrics and their application as programmable materials
-
- Journal:
- Programmable Materials / Volume 2 / 2024
- Published online by Cambridge University Press:
- 23 May 2024, e5
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Possibilities of the X-ray diffraction data processing method for detecting reflections with intensity below the background noise component
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 22 May 2024, pp. 132-143
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Perspectives for extraordinary elastic-wave control in non-Hermitian meta-structures
- Part of
-
- Journal:
- Programmable Materials / Volume 2 / 2024
- Published online by Cambridge University Press:
- 20 May 2024, e4
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure of alectinib hydrochloride Type I, C30H35N4O2Cl
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 20 May 2024, pp. 170-175
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure of rilpivirine hydrochloride, N6H19C22Cl
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 20 May 2024, pp. 151-158
-
- Article
- Export citation
Crystal structures and X-ray powder diffraction data for AAlGe2O6 synthetic leucite analogs (A = K, Rb, Cs)
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 16 May 2024, pp. 162-169
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Leucites are tetrahedrally coordinated silicate framework structures with some of the silicon framework cations that are partially replaced by divalent or trivalent cations. These structures have general formulae A2BSi5O12 and ACSi2O6, where A is a monovalent alkali metal cation, B is a divalent cation, and C is a trivalent cation. There are also leucite analogs with analogous tetrahedrally coordinated germanate framework structures. These have general formulae A2BGe5O12 and ACGe2O6. In this paper, the Rietveld refinements of three synthetic Ge-leucite analogs with stoichiometries of AAlGe2O6 (A = K, Rb, Cs) are discussed. KAlGe2O6 is I41/a tetragonal and is isostructural with KAlSi2O6. RbAlGe2O6 and CsAlGe2O6 are
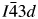 $I\bar{4}3d$ cubic and are isostructural with KBSi2O6.
$I\bar{4}3d$ cubic and are isostructural with KBSi2O6.
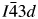
NIST Workshop: Integrating Crystallographic and Computational Approaches to Carbon-Capture Materials for the Mitigation of Climate Change (October 31–November 1, 2023)
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 15 May 2024, pp. 111-114
-
- Article
- Export citation
Crystal structure of valbenazine, C24H38N2O4
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 06 May 2024, pp. 176-181
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
