Refine search
Actions for selected content:
106116 results in Materials Science
Determination of two structures of the solvent 3-hydroxypropionitrile crystallized at low temperatures
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 01 March 2024, pp. 1-11
-
- Article
- Export citation
Crystal structure of ractopamine hydrochloride, C18H24NO3Cl
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 29 February 2024, pp. 94-104
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Simple preparation of specimens for X-ray powder diffraction analysis of radioactive materials: an illustrative example on irradiated granite
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 29 February 2024, pp. 41-46
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure and synchrotron X-ray powder reference pattern for the porous pillared cyanonickelate, Ni(3-amino-4,4′-bipyridine)[Ni(CN)4]
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 29 February 2024, pp. 20-28
-
- Article
- Export citation
Proposed crystal structure of carbadox, C11H10N4O4
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 29 February 2024, pp. 82-93
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure of indacaterol hydrogen maleate (C24H29N2O3)(HC4H2O4)
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 29 February 2024, pp. 76-81
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

How do we grow a Biodesigner?
-
- Journal:
- Research Directions: Biotechnology Design / Volume 2 / 2024
- Published online by Cambridge University Press:
- 28 February 2024, e2
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Crystal structure of anthraquinone-2-carboxylic acid, C15H8O4
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 19 February 2024, pp. 29-35
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Puzzle-shaped cells and the mechanical response of tobacco (Nicotiana tabacum L.) seed coats
- Part of
-
- Journal:
- Programmable Materials / Volume 2 / 2024
- Published online by Cambridge University Press:
- 16 February 2024, e1
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Can we accurately bridge neurobiology to brain-inspired AGI to effectively emulate the human brain?
-
- Journal:
- Research Directions: Bioelectronics / Volume 2 / 2024
- Published online by Cambridge University Press:
- 12 February 2024, e1
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
PDJ volume 38 issue 4 Cover and Back matter
-
- Journal:
- Powder Diffraction / Volume 38 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 08 February 2024, p. b1
-
- Article
-
- You have access
- Export citation
Calendar of short courses and workshops
-
- Journal:
- Powder Diffraction / Volume 38 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 08 February 2024, p. 259
-
- Article
-
- You have access
- HTML
- Export citation
Experimental electron density distribution of KZnB3O6 constructed by maximum-entropy method
-
- Journal:
- Powder Diffraction / Volume 38 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 08 February 2024, pp. 233-239
-
- Article
- Export citation
PDJ volume 38 issue 4 Cover and Front matter
-
- Journal:
- Powder Diffraction / Volume 38 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 08 February 2024, pp. f1-f4
-
- Article
-
- You have access
- Export citation
Adsorption of Ar into zeolite Al-MFI (NH4)
-
- Journal:
- Powder Diffraction / Volume 39 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 08 February 2024, pp. 12-19
-
- Article
- Export citation
Paper waste grown as a biocalcified foam: perspectives from a bacterial and design viewpoint
-
- Journal:
- Research Directions: Biotechnology Design / Volume 2 / 2024
- Published online by Cambridge University Press:
- 10 January 2024, e1
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Provable observation noise robustness for neural network control systems
-
- Journal:
- Research Directions: Cyber-Physical Systems / Volume 2 / 2024
- Published online by Cambridge University Press:
- 08 January 2024, e1
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Neural networks are vulnerable to adversarial perturbations: slight changes to inputs that can result in unexpected outputs. In neural network control systems, these inputs are often noisy sensor readings. In such settings, natural sensor noise – or an adversary who can manipulate them – may cause the system to fail. In this paper, we introduce the first technique to provably compute the minimum magnitude of sensor noise that can cause a neural network control system to violate a safety property from a given initial state. Our algorithm constructs a tree of possible successors with increasing noise until a specification is violated. We build on open-loop neural network verification methods to determine the least amount of noise that could change actions at each step of a closed-loop execution. We prove that this method identifies the unsafe trajectory with the least noise that leads to a safety violation. We evaluate our method on four systems: the Cart Pole and LunarLander environments from OpenAI gym, an aircraft collision avoidance system based on a neural network compression of ACAS Xu, and the SafeRL Aircraft Rejoin scenario. Our analysis produces unsafe trajectories where deviations under
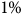 $1{\rm{\% }}$ of the sensor noise range make the systems behave erroneously.
$1{\rm{\% }}$ of the sensor noise range make the systems behave erroneously.

Intelligent Metasurface Sensors
-
- Published online:
- 20 December 2023
- Print publication:
- 01 February 2024
-
- Element
- Export citation
Flow charts as a method to transfer self-sealing from plant models into programmable materials and related challenges
-
- Journal:
- Programmable Materials / Volume 1 / 2023
- Published online by Cambridge University Press:
- 18 December 2023, e12
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Integrated Circuit Fabrication
- Science and Technology
-
- Published online:
- 01 December 2023
- Print publication:
- 16 November 2023
-
- Textbook
- Export citation
