Refine search
Actions for selected content:
106117 results in Materials Science
Appendix C - Green's functions and ab initio quantum transport in the Landauer–Büttiker formalism
-
- Book:
- Introduction to Graphene-Based Nanomaterials
- Published online:
- 05 February 2014
- Print publication:
- 23 January 2014, pp 338-357
-
- Chapter
- Export citation
Preface
-
- Book:
- Introduction to Graphene-Based Nanomaterials
- Published online:
- 05 February 2014
- Print publication:
- 23 January 2014, pp xi-xiv
-
- Chapter
- Export citation
2 - Electronic properties of carbon-based nanostructures
-
- Book:
- Introduction to Graphene-Based Nanomaterials
- Published online:
- 05 February 2014
- Print publication:
- 23 January 2014, pp 11-90
-
- Chapter
- Export citation
Appendix A - Electronic structure calculations: the density functional theory (DFT)
-
- Book:
- Introduction to Graphene-Based Nanomaterials
- Published online:
- 05 February 2014
- Print publication:
- 23 January 2014, pp 314-331
-
- Chapter
- Export citation

A new experimental approach for evaluating the mechanical integrity of interfaces between hard coatings and substrates
-
- Journal:
- MRS Communications / Volume 4 / Issue 1 / March 2014
- Published online by Cambridge University Press:
- 22 January 2014, pp. 19-23
- Print publication:
- March 2014
-
- Article
- Export citation
Field Emission from Laser Cut CNT Fibers and Films– CORRIGENDUM
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 4 / 28 February 2014
- Published online by Cambridge University Press:
- 22 January 2014, p. 606
- Print publication:
- 28 February 2014
-
- Article
-
- You have access
- HTML
- Export citation

Lithium oxide solution in chloride melts as a medium to prepare LiCoO2 nanoparticles
-
- Journal:
- MRS Communications / Volume 4 / Issue 1 / March 2014
- Published online by Cambridge University Press:
- 22 January 2014, pp. 15-18
- Print publication:
- March 2014
-
- Article
- Export citation
X-ray powder diffraction data of the antifungal agents, clotrimazole and fluconazole monohydrate
-
- Journal:
- Powder Diffraction / Volume 29 / Issue 3 / September 2014
- Published online by Cambridge University Press:
- 21 January 2014, pp. 289-294
-
- Article
- Export citation
-
Clotrimazole (C22H17ClN2, 1-[(2-chlorophenyl)(diphenyl)methyl]-1H-imidazole) and fluconazole (C13H12F2N6, 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazole-1-yl)propan-2-ol) are two active pharmaceutical ingredients commonly used in the treatment and prevention of superficial and systemic fungal infections. The X-ray powder diffraction data and the unit cell parameters of anhydrous clotrimazole [Triclinic, P
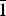 ${\bar 1}$ , a = 8.776(1) Å, b = 10.571(2) Å, c = 10.622(3) Å, α = 114.08(2)°, β = 96.87(2)°, γ = 97.61(2)°, V = 875.2(2) Å3, Z = 2] and monohydrated fluconazole [Triclinic, P
${\bar 1}$ , a = 8.776(1) Å, b = 10.571(2) Å, c = 10.622(3) Å, α = 114.08(2)°, β = 96.87(2)°, γ = 97.61(2)°, V = 875.2(2) Å3, Z = 2] and monohydrated fluconazole [Triclinic, P 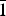 ${\bar 1}$ , a = 5.6353(4) Å, b = 11.753(1) Å, c = 12.326(1) Å, α = 71.220(8)°, β = 79.896(9)°, γ = 84.35(1)°, V = 760.13(9) Å3, Z = 2] are reported.
${\bar 1}$ , a = 5.6353(4) Å, b = 11.753(1) Å, c = 12.326(1) Å, α = 71.220(8)°, β = 79.896(9)°, γ = 84.35(1)°, V = 760.13(9) Å3, Z = 2] are reported.
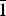

UV photo-chlorination and -bromination of single-walled carbon nanotubes
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 2 / 28 January 2014
- Published online by Cambridge University Press:
- 21 January 2014, pp. 239-246
- Print publication:
- 28 January 2014
-
- Article
- Export citation

Graphene synthesis and application for solar cells
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 3 / 14 February 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. 299-319
- Print publication:
- 14 February 2014
-
- Article
-
- You have access
- HTML
- Export citation
An energy analysis of nanovoid nucleation in nanocrystalline materials with grain boundary sliding accommodations
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 2 / 28 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. 277-287
- Print publication:
- 28 January 2014
-
- Article
- Export citation
Bulge fatigue testing of freestanding and supported gold films
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 2 / 28 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. 267-276
- Print publication:
- 28 January 2014
-
- Article
- Export citation
Effect of microwave processes on the energy-storage properties of barium strontium titanate glass ceramics
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 2 / 28 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. 288-293
- Print publication:
- 28 January 2014
-
- Article
- Export citation

Effect of processing parameters on anodic nanoporous tungsten oxide film structure and porosity for hydrogen detection
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 1 / 14 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. 166-174
- Print publication:
- 14 January 2014
-
- Article
- Export citation
JMR volume 29 issue 1 Cover and Back matter
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 1 / 14 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. b1-b4
- Print publication:
- 14 January 2014
-
- Article
-
- You have access
- Export citation
JMR volume 29 issue 1 Cover and Front matter
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 1 / 14 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. f1-f5
- Print publication:
- 14 January 2014
-
- Article
-
- You have access
- Export citation

Realization of enhanced room temperature ferromagnetism in pure and V-doped ZnO films on TOP functionalization
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 1 / 14 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. 158-165
- Print publication:
- 14 January 2014
-
- Article
- Export citation

The oxidant peroxo method (OPM) as a new alternative for the synthesis of lead-based and bismuth-based oxides
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 1 / 14 January 2014
- Published online by Cambridge University Press:
- 15 January 2014, pp. 131-138
- Print publication:
- 14 January 2014
-
- Article
- Export citation
Barrierless Cu–Ni–Nb thin films on silicon with high thermal stability and low electrical resistivity– ERRATUM
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 4 / 28 February 2014
- Published online by Cambridge University Press:
- 10 January 2014, p. 605
- Print publication:
- 28 February 2014
-
- Article
-
- You have access
- HTML
- Export citation

Characterizing phonon thermal conduction in polycrystalline graphene
-
- Journal:
- Journal of Materials Research / Volume 29 / Issue 3 / 14 February 2014
- Published online by Cambridge University Press:
- 10 January 2014, pp. 362-372
- Print publication:
- 14 February 2014
-
- Article
- Export citation
