Refine search
Actions for selected content:
238502 results in Physics and Astronomy

Nanoscale surfactant transport: bridging molecular and continuum models
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 15 April 2025, A18
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Rapidly yawing spheroids in viscous shear flow: emergent loss of symmetry
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 15 April 2025, A27
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Asymptotic drag reduction states in turbulent Taylor vortex flow of dilute polymeric solutions: interplay between large-scale structures and polymer chains dynamics
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 15 April 2025, A24
-
- Article
- Export citation
-
Direct numerical simulations in a low-curvature viscoelastic turbulent Taylor vortex flow, with Reynolds numbers ranging from 1500 to 8000 and maximum chain extensibility (
 $L$) from 50 to 200, reveal a maximum drag reduction (MDR) asymptote. Compared with the classical MDR observed in planar wall-bounded shear flows, that is, drag reduction (DR) is
$L$) from 50 to 200, reveal a maximum drag reduction (MDR) asymptote. Compared with the classical MDR observed in planar wall-bounded shear flows, that is, drag reduction (DR) is  $\sim -80\, \%$, this MDR state achieves only moderate levels of DR (
$\sim -80\, \%$, this MDR state achieves only moderate levels of DR ( $\sim -60\,\%$). This is due to the existence of large-scale structures (LSSs). A careful examination of the flow structures reveals that the polymer–turbulence interaction suppresses small-scale vortices and stabilizes the LSSs. These structural changes in turn lead to a reduction of Reynolds stress, and consequently to a DR flow state. Although Reynolds stress does not vanish as observed in classical MDR states, the small-scale vortices that heavily populate the near-wall region are also almost completely eliminated in this flow state. Concurrently, significant polymer stresses develop as a consequence of the interaction between polymer chains and LSSs that partially offset the magnitude of DR, leading to MDR asymptotes with moderate levels of DR. Moreover, we demonstrate that polymer deformation, i.e. deviation from the equilibrium state, is directly correlated with the LSSs dynamics, while the polymer deformation fluctuation displays a universal property in the MDR state. Hence, it is not surprising that the extent of DR exhibits a non-monotonic dependence on the maximum chain extensibility. Specifically, the variation in
$\sim -60\,\%$). This is due to the existence of large-scale structures (LSSs). A careful examination of the flow structures reveals that the polymer–turbulence interaction suppresses small-scale vortices and stabilizes the LSSs. These structural changes in turn lead to a reduction of Reynolds stress, and consequently to a DR flow state. Although Reynolds stress does not vanish as observed in classical MDR states, the small-scale vortices that heavily populate the near-wall region are also almost completely eliminated in this flow state. Concurrently, significant polymer stresses develop as a consequence of the interaction between polymer chains and LSSs that partially offset the magnitude of DR, leading to MDR asymptotes with moderate levels of DR. Moreover, we demonstrate that polymer deformation, i.e. deviation from the equilibrium state, is directly correlated with the LSSs dynamics, while the polymer deformation fluctuation displays a universal property in the MDR state. Hence, it is not surprising that the extent of DR exhibits a non-monotonic dependence on the maximum chain extensibility. Specifically, the variation in  $L$ alters the incoherent and coherent angular momentum transport by small- and large-scale flow structures, respectively. To that end, the most DR flow state occurs at a moderate value
$L$ alters the incoherent and coherent angular momentum transport by small- and large-scale flow structures, respectively. To that end, the most DR flow state occurs at a moderate value  $L=100$. Overall, this study further supports the universal property of polymer-induced asymptotic states in wall-bounded turbulence and paves the way for mechanistic understanding of drag modification that arises from the interaction of polymers with small- and large-scale flow structures.
$L=100$. Overall, this study further supports the universal property of polymer-induced asymptotic states in wall-bounded turbulence and paves the way for mechanistic understanding of drag modification that arises from the interaction of polymers with small- and large-scale flow structures.





Estimating the noise budget and system noise levels in closed-loop Doppler tracking of ESA’s Mars Express with VLBI radio telescopes
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 42 / 2025
- Published online by Cambridge University Press:
- 15 April 2025, e054
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The radio telescopes of the European VLBI Network (EVN) and the University of Tasmania (UTAS) conducted an extensive observation campaign of the European Space Agency’s (ESA) Mars Express (MEX) spacecraft between 2013 and 2020. The campaign, carried out under the Planetary Radio Interferometry and Doppler Experiment (PRIDE) framework, aimed to study interplanetary phase scintillation and assess the noise budget in the closed-loop Doppler observations. The average closed-loop Doppler noise was determined to be approximately 10 mHz at a 10-s integration time, reaffirming the technique’s suitability for radio science experiments. We evaluated how different observational parameters such as the solar elongation, antenna size, and elevation angle impact the Doppler noise. A key part of the analysis involved comparing results from co-located telescopes to investigate system noise effects. Co-located telescopes at both Wettzell and Hobart provided highly consistent results, with any deviations serving as diagnostic tools to identify station-dependent issues. Additionally, the use of phase calibration tones during spacecraft tracking showed that the instrumental noise contribution is of the order of 5
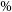 $\%$ of the total noise. This study provides a detailed noise budget for closed-loop Doppler observations with VLBI telescopes while emphasizing the effectiveness of the co-location method in isolating system-level noise. These findings are important for optimizing future radio science and VLBI tracking missions using stations outside the the Deep Space Network (DSN) and European Space Tracking (ESTRACK) network.
$\%$ of the total noise. This study provides a detailed noise budget for closed-loop Doppler observations with VLBI telescopes while emphasizing the effectiveness of the co-location method in isolating system-level noise. These findings are important for optimizing future radio science and VLBI tracking missions using stations outside the the Deep Space Network (DSN) and European Space Tracking (ESTRACK) network.

A hybrid numerical model for the collective motion of fish groups
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A26
-
- Article
- Export citation

Flow separation, instability and transition to turbulence on a cambered airfoil at Reynolds number 20 000
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A9
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The flow over a cambered NACA 65(1)–412 airfoil at
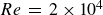 $Re\,=\,2\times 10^4$ is described based on a high-order direct numerical simulation. Simulations are run over a range of angles of attack,
$Re\,=\,2\times 10^4$ is described based on a high-order direct numerical simulation. Simulations are run over a range of angles of attack, 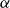 $\alpha$, where a number of instabilities in the unsteady, three-dimensional flow field are identified. The balance and competing effects of these instabilities are responsible for significant and abrupt (with respect to
$\alpha$, where a number of instabilities in the unsteady, three-dimensional flow field are identified. The balance and competing effects of these instabilities are responsible for significant and abrupt (with respect to 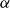 $\alpha$) changes in flow regime, with measurable consequences in time-averaged, integrated force coefficients, and in the far-wake footprint. At low
$\alpha$) changes in flow regime, with measurable consequences in time-averaged, integrated force coefficients, and in the far-wake footprint. At low 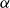 $\alpha$, the flow is strongly influenced by vortex roll-up from the pressure side at the trailing edge. The interaction of this large-scale structure with shear and three-dimensional modal instabilities in the separated shear layer and associated wake region on the suction side, explains the transitions and bifurcations of the the flow states as
$\alpha$, the flow is strongly influenced by vortex roll-up from the pressure side at the trailing edge. The interaction of this large-scale structure with shear and three-dimensional modal instabilities in the separated shear layer and associated wake region on the suction side, explains the transitions and bifurcations of the the flow states as 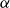 $\alpha$ increases. The transition from a separation at low
$\alpha$ increases. The transition from a separation at low 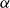 $\alpha$ to reattachment and establishment of a laminar separation bubble at the trailing edge at critical
$\alpha$ to reattachment and establishment of a laminar separation bubble at the trailing edge at critical 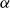 $\alpha$ is driven by instabilities within the separated shear layer that are absent at lower angles. Instabilities of different wavelengths are then shown to pave the path to turbulence in the near wake.
$\alpha$ is driven by instabilities within the separated shear layer that are absent at lower angles. Instabilities of different wavelengths are then shown to pave the path to turbulence in the near wake.
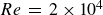
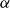
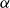
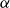
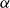
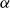

Transient characteristics of fully localised turbulence in transitional channel flow
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, R2
-
- Article
- Export citation
-
The temporal characteristics of fully localised turbulent bands in transitional channel flow remains unclear due to the difficulty in resolving the large length and time scales involved. Here, we tackle this problem by performing statistical lifetime studies in sufficiently large computational domains. The results show signs of stochastic memoryless decay of a fully localised band, suggesting a chaotic-saddle behaviour of the entire band as a coherent entity. Although the mean lifetime of a turbulent band was reported to increase with the band length, our data suggest that it saturates at a certain length. This saturation results in a characteristic lifetime for a fully developed band with a changing length due to the intermittent chipping and decay of turbulence at the upstream end. This memoryless behaviour is observed down to Reynolds number
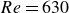 $Re=630$ in our study and we propose that the onset of the memoryless behaviour is in the range of
$Re=630$ in our study and we propose that the onset of the memoryless behaviour is in the range of 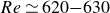 $Re\simeq 620{-}630$. Our data also show that the time it takes for a perturbed flow to enter the saddle, i.e. to start behaving memorylessly, can be thousands of convective time units, which is comparable to the maximum achievable observation time in existing channel set-ups and may pose difficulties for experiments.
$Re\simeq 620{-}630$. Our data also show that the time it takes for a perturbed flow to enter the saddle, i.e. to start behaving memorylessly, can be thousands of convective time units, which is comparable to the maximum achievable observation time in existing channel set-ups and may pose difficulties for experiments.
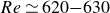
Percent-level timing of reionisation: Self-consistent, implicit-likelihood inference from XQR-30+ Lyα forest data
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 42 / 2025
- Published online by Cambridge University Press:
- 14 April 2025, e049
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The Lyman alpha (Ly
 $\alpha$) forest in the spectra of
$\alpha$) forest in the spectra of 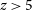 $z\gt5$ quasars provides a powerful probe of the late stages of the epoch of reionisation (EoR). With the recent advent of exquisite datasets such as XQR-30, many models have struggled to reproduce the observed large-scale fluctuations in the Ly
$z\gt5$ quasars provides a powerful probe of the late stages of the epoch of reionisation (EoR). With the recent advent of exquisite datasets such as XQR-30, many models have struggled to reproduce the observed large-scale fluctuations in the Ly $\alpha$ opacity. Here we introduce a Bayesian analysis framework that forward-models large-scale lightcones of intergalactic medium (IGM) properties and accounts for unresolved sub-structure in the Ly
$\alpha$ opacity. Here we introduce a Bayesian analysis framework that forward-models large-scale lightcones of intergalactic medium (IGM) properties and accounts for unresolved sub-structure in the Ly $\alpha$ opacity by calibrating to higher-resolution hydrodynamic simulations. Our models directly connect physically intuitive galaxy properties with the corresponding IGM evolution, without having to tune ‘effective’ parameters or calibrate out the mean transmission. The forest data, in combination with UV luminosity functions and the CMB optical depth, are able to constrain global IGM properties at percent level precision in our fiducial model. Unlike many other works, we recover the forest observations without invoking a rapid drop in the ionising emissivity from
$\alpha$ opacity by calibrating to higher-resolution hydrodynamic simulations. Our models directly connect physically intuitive galaxy properties with the corresponding IGM evolution, without having to tune ‘effective’ parameters or calibrate out the mean transmission. The forest data, in combination with UV luminosity functions and the CMB optical depth, are able to constrain global IGM properties at percent level precision in our fiducial model. Unlike many other works, we recover the forest observations without invoking a rapid drop in the ionising emissivity from 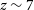 $z\sim7$ to 5.5, which we attribute to our sub-grid model for recombinations. In this fiducial model, reionisation ends at
$z\sim7$ to 5.5, which we attribute to our sub-grid model for recombinations. In this fiducial model, reionisation ends at 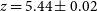 $z=5.44\pm0.02$ and the EoR mid-point is at
$z=5.44\pm0.02$ and the EoR mid-point is at 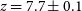 $z=7.7\pm0.1$. The ionising escape fraction increases towards faint galaxies, showing a mild redshift evolution at fixed UV magnitude,
$z=7.7\pm0.1$. The ionising escape fraction increases towards faint galaxies, showing a mild redshift evolution at fixed UV magnitude, 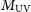 $M_\textrm{UV}$. Half of the ionising photons are provided by galaxies fainter than
$M_\textrm{UV}$. Half of the ionising photons are provided by galaxies fainter than 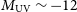 $M_\textrm{UV} \sim -12$, well below direct detection limits of optical/NIR instruments including
$M_\textrm{UV} \sim -12$, well below direct detection limits of optical/NIR instruments including 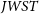 $\textit{ JWST}$. We also show results from an alternative galaxy model that does not allow for a redshift evolution in the ionising escape fraction. Despite being decisively disfavoured by the Bayesian evidence, the posterior of this model is in qualitative agreement with that from our fiducial model. We caution, however, that our conclusions regarding the early stages of the EoR and which sources reionised the Universe are more model-dependent.
$\textit{ JWST}$. We also show results from an alternative galaxy model that does not allow for a redshift evolution in the ionising escape fraction. Despite being decisively disfavoured by the Bayesian evidence, the posterior of this model is in qualitative agreement with that from our fiducial model. We caution, however, that our conclusions regarding the early stages of the EoR and which sources reionised the Universe are more model-dependent.

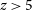


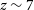
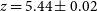
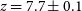
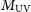
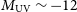
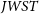

Leveraging initial conditions memory for modelling Rayleigh–Taylor turbulence
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A17
-
- Article
- Export citation

Solute transport due to periodic loading in a soft porous material
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A15
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Harmonic decomposition of forces and estimates of reduced mean flow in jackets subjected to waves and current
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A7
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Two-layer formulation for long-runout turbidity currents: theory and bypass flow case
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A19
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Roughness-induced boundary-layer instability beneath internal solitary waves
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A21
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
We investigate the effects of bottom roughness on bottom boundary-layer (BBL) instability beneath internal solitary waves (ISWs) of depression. Applying both two-dimensional (2-D) numerical simulations and linear stability theory, an extensive parametric study explores the effect of the Reynolds number, pressure gradient, roughness (periodic bump) height
 $h_b$ and roughness wavelength
$h_b$ and roughness wavelength  $\lambda _b$ on BBL instability. The simulations show that small
$\lambda _b$ on BBL instability. The simulations show that small  $h_b$, comparable to that of laboratory-flume materials (
$h_b$, comparable to that of laboratory-flume materials ( $\sim$100 times less than the thickness of the viscous sublayer
$\sim$100 times less than the thickness of the viscous sublayer  $\delta _v$), can destabilize the BBL and trigger vortex shedding at critical Reynolds numbers much lower than what occurs for numerically smooth surfaces. We identify two mechanisms of vortex shedding, depending on
$\delta _v$), can destabilize the BBL and trigger vortex shedding at critical Reynolds numbers much lower than what occurs for numerically smooth surfaces. We identify two mechanisms of vortex shedding, depending on  $h_b/\delta _v$. For
$h_b/\delta _v$. For  $h_b/\delta _v \gtrapprox 1$, vortices are forced directly by local flow separation in the lee of each bump. Conversely, for
$h_b/\delta _v \gtrapprox 1$, vortices are forced directly by local flow separation in the lee of each bump. Conversely, for  $h_b/\delta _v \lessapprox 10^{-1}$ the roughness seeds perturbations in the BBL, which are amplified by the BBL flow. Roughness wavelengths close to those associated with the most unstable BBL mode, as predicted by linear instability theory, are preferentially amplified. This resonant amplification nature of the BBL flow, beneath ISWs, is consistent with what occurs in a BBL driven by surface solitary waves and by periodic monochromatic waves. Using the
$h_b/\delta _v \lessapprox 10^{-1}$ the roughness seeds perturbations in the BBL, which are amplified by the BBL flow. Roughness wavelengths close to those associated with the most unstable BBL mode, as predicted by linear instability theory, are preferentially amplified. This resonant amplification nature of the BBL flow, beneath ISWs, is consistent with what occurs in a BBL driven by surface solitary waves and by periodic monochromatic waves. Using the  $N$-factor method for Tollmien–Schlichting waves, we propose an analogy between the roughness height and seed noise required to trigger instability. Including surface roughness, or more generally an appropriate level of seed noise, reconciles the discrepancies between the vortex-shedding threshold observed in the laboratory versus that predicted by otherwise smooth-bottomed 2-D spectral simulations.
$N$-factor method for Tollmien–Schlichting waves, we propose an analogy between the roughness height and seed noise required to trigger instability. Including surface roughness, or more generally an appropriate level of seed noise, reconciles the discrepancies between the vortex-shedding threshold observed in the laboratory versus that predicted by otherwise smooth-bottomed 2-D spectral simulations.










Layer formation in double-diffusive convection diagnosed in sorted buoyancy coordinates
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, R3
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Double-diffusive convection can arise when the fluid density is set by multiple species which diffuse at different rates. Different flow regimes are possible depending on the distribution of the diffusing species, including salt fingering and diffusive convection. Flows arising from diffusive convection commonly exhibit step-like density profiles with sharp density interfaces separated by well-mixed layers. The formation of density layers is also seen in stratified turbulence, where a framework based on sorted density coordinates (Winters & D’Asaro 1996 J. Fluid Mech. 317, 179–193) has been used to diagnose layer formation (Zhou et al. 2017 J. Fluid Mech. 823, 198–229; Taylor & Zhou 2017 J. Fluid Mech. 823, R5). In this framework, the evolution of the sorted density profile can be expressed solely in terms of the eddy diffusivity,
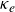 $\kappa _e$. Here, we use the same framework to diagnose layer formation in two-dimensional simulations of double-diffusive convection. We show that
$\kappa _e$. Here, we use the same framework to diagnose layer formation in two-dimensional simulations of double-diffusive convection. We show that 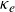 $\kappa _e$ is negative everywhere, with the antidiffusive effect strongest in ‘well-mixed’ layers where a positive diffusion coefficient may be expected. By considering a decomposition of the budget of the square of the Brunt-Väisälä frequency
$\kappa _e$ is negative everywhere, with the antidiffusive effect strongest in ‘well-mixed’ layers where a positive diffusion coefficient may be expected. By considering a decomposition of the budget of the square of the Brunt-Väisälä frequency 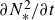 $\partial N^2_*/\partial t$, we demonstrate that the density layers are maintained by fundamentally different processes than in single-component stratified turbulence. In more complicated flows where stratified turbulence and double-diffusive convection can coexist, this framework could provide a method to distinguish between the mechanisms responsible for generating density layers.
$\partial N^2_*/\partial t$, we demonstrate that the density layers are maintained by fundamentally different processes than in single-component stratified turbulence. In more complicated flows where stratified turbulence and double-diffusive convection can coexist, this framework could provide a method to distinguish between the mechanisms responsible for generating density layers.
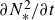

Transient segregation of different density granular mixtures
-
- Journal:
- Journal of Fluid Mechanics / Volume 1008 / 10 April 2025
- Published online by Cambridge University Press:
- 14 April 2025, A53
-
- Article
- Export citation
-
We study time-dependent density segregation of granular mixtures flowing over an inclined plane. Discrete element method (DEM) simulations in a periodic box are performed for granular mixtures of same size and different density particles flowing under the influence of gravity. In addition, a continuum model is developed to solve the momentum balance equations along with the species transport equation by accounting for the inter-coupling of segregation and rheology. The particle force-based density segregation theory has been used along with the
 $\mu {-}I$ rheology to predict evolution of flow properties with time for binary and multicomponent mixtures. The effect of particle arrangements on the transient evolution of flow properties for three different initial configurations is investigated using both continuum and DEM simulations. Continuum predictions for various flow properties of interest such as species concentration, velocity, pressure and shear stress at different time instants are compared with DEM simulations. The results from the discrete and continuum models are found to be in good agreement with each other for well-mixed and heavy-near-base initial configurations. Kinetic theory-based predictions of segregation evolution, however, show good quantitative agreement only for the heavy-near-base configuration with a much slower evolution for the well-mixed case. Interestingly, the continuum model is unable to predict the flow evolution for the light-near-base initial configuration. DEM simulations reveal the presence of an instability driven, quick segregation for this configuration which is not predicted by the one-dimensional model and requires generalisation to three dimensions.
$\mu {-}I$ rheology to predict evolution of flow properties with time for binary and multicomponent mixtures. The effect of particle arrangements on the transient evolution of flow properties for three different initial configurations is investigated using both continuum and DEM simulations. Continuum predictions for various flow properties of interest such as species concentration, velocity, pressure and shear stress at different time instants are compared with DEM simulations. The results from the discrete and continuum models are found to be in good agreement with each other for well-mixed and heavy-near-base initial configurations. Kinetic theory-based predictions of segregation evolution, however, show good quantitative agreement only for the heavy-near-base configuration with a much slower evolution for the well-mixed case. Interestingly, the continuum model is unable to predict the flow evolution for the light-near-base initial configuration. DEM simulations reveal the presence of an instability driven, quick segregation for this configuration which is not predicted by the one-dimensional model and requires generalisation to three dimensions.


Reverse capillary trapping and self-removal of non-aqueous fluid from dead-end structures by nanoparticle suspension
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A14
-
- Article
- Export citation

Role of finite extensibility on the centre-mode instability in viscoelastic channel flow
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A28
-
- Article
- Export citation
-
A purely elastic linear instability was recently reported for viscoelastic plane Poiseuille flow in the limit of ultra-dilute (solvent to solution viscosity ratio
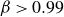 $\beta \gt 0.99$), highly elastic (Weissenberg number
$\beta \gt 0.99$), highly elastic (Weissenberg number 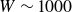 $W \sim 1000$) polymer solutions, within the framework of the Oldroyd-B model (Khalid et al., Phys. Rev. Lett., vol. 127, 2021, pp. 134–502). This is the first instance of a purely elastic instability in a strictly rectilinear shearing flow, with the phase speed of the unstable ‘centre mode’ being close to the base-state maximum velocity at the channel centreline. Subsequently, Buza, Page and Kerswell (J. Fluid Mech., vol. 940, 2022, A11) have shown, using the FENE-P model, that the centre-mode instability persists down to moderate elasticities (
$W \sim 1000$) polymer solutions, within the framework of the Oldroyd-B model (Khalid et al., Phys. Rev. Lett., vol. 127, 2021, pp. 134–502). This is the first instance of a purely elastic instability in a strictly rectilinear shearing flow, with the phase speed of the unstable ‘centre mode’ being close to the base-state maximum velocity at the channel centreline. Subsequently, Buza, Page and Kerswell (J. Fluid Mech., vol. 940, 2022, A11) have shown, using the FENE-P model, that the centre-mode instability persists down to moderate elasticities (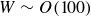 $W \sim O (100)$), the reduction in threshold evidently due to the finite extensibility of the polymer molecules. In this work, we augment this latter finding and provide a comprehensive account of the effect of finite extensibility on the centre-mode instability in viscoelastic channel flow, using the FENE-P and FENE-CR models, in both the absence and presence of fluid inertia. In both these models, finite extensibility causes a decrease in the polymer relaxation time at high shear rates, and the resulting weakening of elastic stresses would seem to indicate a stabilising effect. The latter trend has been demonstrated by earlier analyses of hoop-stress-driven instabilities in curvilinear flows, and is indeed borne out for the FENE-CR case, where finite extensibility has a largely stabilising influence on the centre-mode instability. In stark contrast, for the FENE-P model, finite extensibility plays a dual role – a stabilising one at lower values of the elasticity number
$W \sim O (100)$), the reduction in threshold evidently due to the finite extensibility of the polymer molecules. In this work, we augment this latter finding and provide a comprehensive account of the effect of finite extensibility on the centre-mode instability in viscoelastic channel flow, using the FENE-P and FENE-CR models, in both the absence and presence of fluid inertia. In both these models, finite extensibility causes a decrease in the polymer relaxation time at high shear rates, and the resulting weakening of elastic stresses would seem to indicate a stabilising effect. The latter trend has been demonstrated by earlier analyses of hoop-stress-driven instabilities in curvilinear flows, and is indeed borne out for the FENE-CR case, where finite extensibility has a largely stabilising influence on the centre-mode instability. In stark contrast, for the FENE-P model, finite extensibility plays a dual role – a stabilising one at lower values of the elasticity number 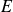 $E$, but, surprisingly, a destabilising one at higher
$E$, but, surprisingly, a destabilising one at higher 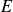 $E$ values. Further, the centre-mode instability is predicted over a significantly larger domain of the
$E$ values. Further, the centre-mode instability is predicted over a significantly larger domain of the 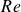 $Re$–
$Re$–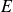 $E$–
$E$–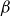 $\beta$ parameter space, compared to the Oldroyd-B model, making it more amenable to experimental observations.
$\beta$ parameter space, compared to the Oldroyd-B model, making it more amenable to experimental observations.
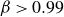
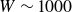
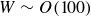
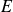
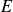
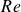
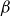

Insights into instability modes of supersonic square jets
-
- Journal:
- Journal of Fluid Mechanics / Volume 1009 / 10 May 2025
- Published online by Cambridge University Press:
- 14 April 2025, A13
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
On the stability of the m = 1 rigid ballooning mode in a mirror trap with high-beta sloshing ions
- Part of
-
- Journal:
- Journal of Plasma Physics / Volume 91 / Issue 2 / April 2025
- Published online by Cambridge University Press:
- 14 April 2025, E54
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Stability of the ‘rigid’ ($m = 1$
 ) ballooning mode in a mirror axisymmetric trap is studied for the case of oblique neutral beam injection (NBI), which creates an anisotropic population of fast sloshing ions. Since small-scale modes with azimuthal numbers $m>1$
) ballooning mode in a mirror axisymmetric trap is studied for the case of oblique neutral beam injection (NBI), which creates an anisotropic population of fast sloshing ions. Since small-scale modes with azimuthal numbers $m>1$ in long thin (paraxial) mirror traps are easily stabilized by finite-Larmor-radius (FLR) effects, suppression of the rigid ballooning and flute modes would mean stabilization of all magnetohydrodynamic (MHD) modes, with the exception of the mirror and firehose disturbances, which are intensively studied in geophysics, but have not yet been identified in mirror traps. Large-scale ballooning mode can, in principle, be suppressed either by the lateral perfectly conducting wall, or by the end MHD anchors such as the cusp, by biased limiters or by a combination of these two methods. The effects of the wall shape, vacuum gap width between the plasma column and the lateral wall, angle of oblique NBI, radial profile of the plasma pressure and axial profile of the vacuum magnetic field are studied. It is confirmed that the lateral conducting wall still creates the upper stability zone, where the ratio $\beta$
in long thin (paraxial) mirror traps are easily stabilized by finite-Larmor-radius (FLR) effects, suppression of the rigid ballooning and flute modes would mean stabilization of all magnetohydrodynamic (MHD) modes, with the exception of the mirror and firehose disturbances, which are intensively studied in geophysics, but have not yet been identified in mirror traps. Large-scale ballooning mode can, in principle, be suppressed either by the lateral perfectly conducting wall, or by the end MHD anchors such as the cusp, by biased limiters or by a combination of these two methods. The effects of the wall shape, vacuum gap width between the plasma column and the lateral wall, angle of oblique NBI, radial profile of the plasma pressure and axial profile of the vacuum magnetic field are studied. It is confirmed that the lateral conducting wall still creates the upper stability zone, where the ratio $\beta$ of the plasma pressure to the pressure of vacuum magnetic field exceeds the second critical value $\beta_{\text{cr2}}$
of the plasma pressure to the pressure of vacuum magnetic field exceeds the second critical value $\beta_{\text{cr2}}$ , $\beta >\beta _{\text {cr2}}$
, $\beta >\beta _{\text {cr2}}$ . However, in many cases the upper zone is clamped from above by mirror instability. When the lateral wall is combined with end MHD anchors, a lower stability zone $\beta <\beta _{\text {cr}1}$
. However, in many cases the upper zone is clamped from above by mirror instability. When the lateral wall is combined with end MHD anchors, a lower stability zone $\beta <\beta _{\text {cr}1}$ appears, where $\beta$
appears, where $\beta$ is below the first critical value $\beta_{\text{cr1}}$
is below the first critical value $\beta_{\text{cr1}}$ . These two zones can overlap in the case of a sufficiently smooth radial pressure profile, and/or a sufficiently low mirror ratio and/or a sufficiently narrow vacuum gap between the plasma column and the lateral wall. However, even in this case, the range of permissible values of beta is limited from above by the threshold of mirror instability $\beta_{\text{mm}}$
. These two zones can overlap in the case of a sufficiently smooth radial pressure profile, and/or a sufficiently low mirror ratio and/or a sufficiently narrow vacuum gap between the plasma column and the lateral wall. However, even in this case, the range of permissible values of beta is limited from above by the threshold of mirror instability $\beta_{\text{mm}}$ , so that $\beta <\beta _{\text {mm}}<1$
, so that $\beta <\beta _{\text {mm}}<1$ , in contrast to the case of transversal NBI, when neutral beams are injected perpendicularly to the magnetic field.
, in contrast to the case of transversal NBI, when neutral beams are injected perpendicularly to the magnetic field.










Effects of angular scattering and H+p, H+H collisions on the properties of interstellar atoms in the heliosphere
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 42 / 2025
- Published online by Cambridge University Press:
- 14 April 2025, e045
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Interstellar hydrogen atoms (H atoms) penetrate into the heliosphere through the region of the solar wind interaction with the interstellar plasma due to their large mean free path. Resonant charge exchange of H atoms with protons has been considered as the main interaction process between the components. In the majority of models, other processes like elastic H-H and H-p collisions are not included. Moreover, it has been assumed that the velocities of the colliding particles remain unchanged during charge exchange. This corresponds to the scattering on the angle of
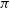 $\pi$ in the centre mass rest frame. The goal of this paper is to explore effects of the elastic H-H and H-p collisions as well as the angular scattering during charge exchange on the distribution of the interstellar atoms in the heliosphere and at its boundary. We present results of simple (and therefore, easily repeatable) kinetic model of the interstellar atom penetration through the region of the solar and interstellar winds interaction into the heliosphere. As a result of the model, we compute the distribution function of the interstellar atoms at different heliospheric distances. Further, this distribution function is used to compute its moments and potentially observable features such as absorption and backscattered spectra in the Lyman-alpha line. Results show that there are differences in the behaviour of the distribution function when considering elastic collisions and the changes in the moments of the distribution achieve 10%. Therefore, in cases where precise calculation of H atom parameters is essential, such as in the modelling of backscattered Lyman-
$\pi$ in the centre mass rest frame. The goal of this paper is to explore effects of the elastic H-H and H-p collisions as well as the angular scattering during charge exchange on the distribution of the interstellar atoms in the heliosphere and at its boundary. We present results of simple (and therefore, easily repeatable) kinetic model of the interstellar atom penetration through the region of the solar and interstellar winds interaction into the heliosphere. As a result of the model, we compute the distribution function of the interstellar atoms at different heliospheric distances. Further, this distribution function is used to compute its moments and potentially observable features such as absorption and backscattered spectra in the Lyman-alpha line. Results show that there are differences in the behaviour of the distribution function when considering elastic collisions and the changes in the moments of the distribution achieve 10%. Therefore, in cases where precise calculation of H atom parameters is essential, such as in the modelling of backscattered Lyman- $\alpha$ emission, elastic collisions must be considered.
$\alpha$ emission, elastic collisions must be considered.