Refine listing
Actions for selected content:
1417513 results in Open Access
Customized Spectral Libraries for Effective Mineral Exploration: Mining National Mineral Collections
-
- Journal:
- Clays and Clay Minerals / Volume 66 / Issue 3 / June 2018
- Published online by Cambridge University Press:
- 01 January 2024, pp. 297-314
-
- Article
- Export citation
In situ X-ray Diffraction Study of the Swelling of Montmorillonite as Affected by Exchangeable Cations and Temperature
-
- Journal:
- Clays and Clay Minerals / Volume 59 / Issue 2 / April 2011
- Published online by Cambridge University Press:
- 01 January 2024, pp. 165-175
-
- Article
- Export citation
Influence of Mechanical Compaction and Clay Mineral Diagenesis on the Microfabric and Pore-Scale Properties of Deep-Water Gulf of Mexico Mudstones
-
- Journal:
- Clays and Clay Minerals / Volume 54 / Issue 4 / August 2006
- Published online by Cambridge University Press:
- 01 January 2024, pp. 500-514
-
- Article
- Export citation
Benzene Vapor Sorption by Organobentonites From Ambient Air
-
- Journal:
- Clays and Clay Minerals / Volume 50 / Issue 4 / August 2002
- Published online by Cambridge University Press:
- 01 January 2024, pp. 421-427
-
- Article
- Export citation
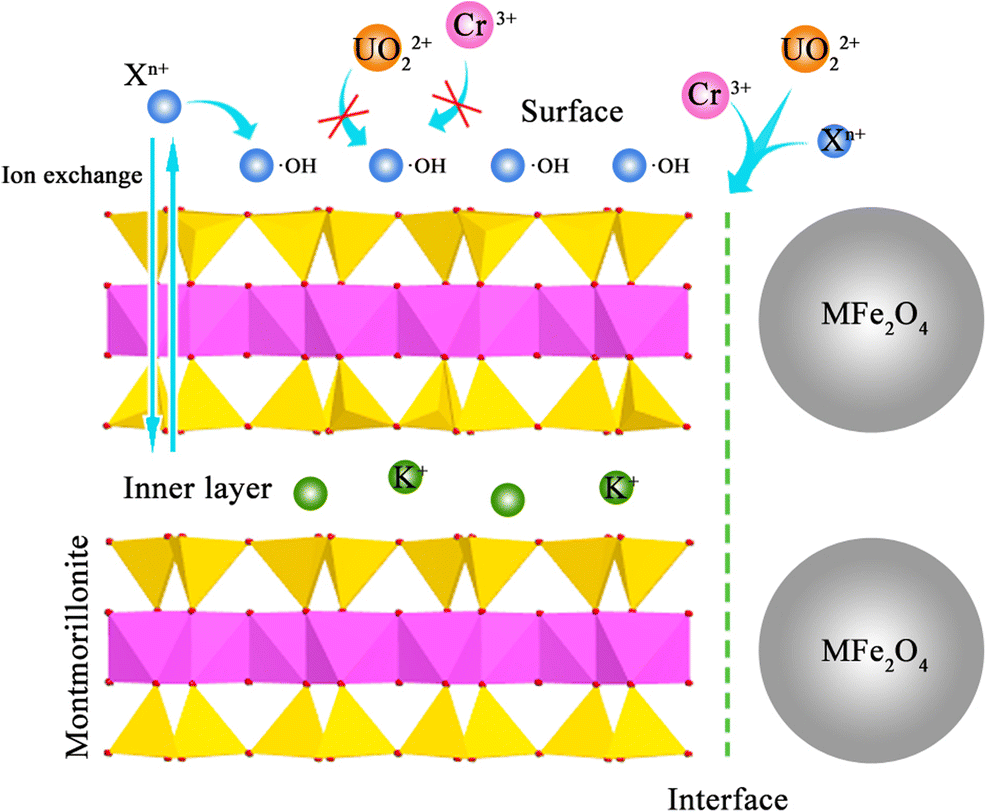
Competitive Adsorption of Uranyl and Toxic Trace Metal Ions at MFe2O4-montmorillonite (M = Mn, Fe, Zn, Co, or Ni) Interfaces
-
- Journal:
- Clays and Clay Minerals / Volume 67 / Issue 4 / August 2019
- Published online by Cambridge University Press:
- 01 January 2024, pp. 291-305
-
- Article
- Export citation
-
Adsorption of uranyl (UO22+) ions to mineral surfaces is a potentially effective method for removing this hazardous metal from water, but other toxic trace metal ions (Xn+: Rb+, Sr2+, Cr3+, Mn2+, Ni2+, Zn2+, Cd2+) in uraniferous wastewaters compete with UO22+ for adsorption sites and thus may diminish the capacity of adsorbents to sequester UO22+. A better understanding of competitive adsorption among these metal ions and the development of better adsorbents are, therefore, of critical importance. The purpose of the present study was to synthesize and characterize magnetic adsorbents, consisting of MFe2O4 (M = Mn, Fe, Zn, Co, or Ni) nanoparticles synthesized on montmorillonite (Mnt) edge sites, and to investigate their use as adsorbents for UO22+, including competitive adsorption with trace metal ions. Selective adsorption was studied using Langmuir, Freundlich, and Dubinin-Radushkevich isotherms, and the results showed that Xn+ ions were adsorbed primarily on MFe2O4-montmorillonite surfaces, and the UO22+ ions were adsorbed on the interfaces between montmorillonite edge surfaces and MFe2O4 nanoparticles. Using the Freundlich model, the interface adsorption capacity of UO22+ reached 25.1 mg·g–1 in mixed solution. Further, the UO22+ and Cr3+ ions had a redox reaction on the interfaces with synergistic adsorption. Herein, the adsorption capacity of Cr3+ was 60.2 mg·g–1 using the Freundlich isotherm. The results demonstrated that the MFe2O4-montmorillonite with highly selective adsorption of UO22+ ions is applicable to UO22+ treatment in the presence of toxic trace metal ions.
Graphical abstract

The Role of the Clergy in the Establishment and Consolidation of Pahlavi I (1925–1941)
-
- Journal:
- Iranian Studies / Volume 57 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 29 January 2024, pp. 143-163
- Print publication:
- January 2024
-
- Article
- Export citation
Boron and Lithium Isotopic Signatures of Nanometer-Sized Smectite-Rich Mixed-Layers of Bentonite Beds From Campos Basin (Brazil)
-
- Journal:
- Clays and Clay Minerals / Volume 70 / Issue 1 / February 2022
- Published online by Cambridge University Press:
- 01 January 2024, pp. 72-83
-
- Article
- Export citation
A Molality-Based BET Equation for Modeling the Activity of Water Sorbed on Clay Minerals
-
- Journal:
- Clays and Clay Minerals / Volume 60 / Issue 6 / December 2012
- Published online by Cambridge University Press:
- 01 January 2024, pp. 599-609
-
- Article
- Export citation
A critical examination of safety culture in the superyacht industry
-
- Journal:
- The Journal of Navigation / Volume 77 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 13 September 2024, pp. 100-115
- Print publication:
- January 2024
-
- Article
- Export citation
Arsenic Sorption onto Soils Enriched in Mn and Fe Minerals
-
- Journal:
- Clays and Clay Minerals / Volume 51 / Issue 2 / April 2003
- Published online by Cambridge University Press:
- 01 January 2024, pp. 197-204
-
- Article
- Export citation
Significance of phyllosilicate mineralogy and mineral chemistry in an epithermal environment. Insights from the palai-islica Au-Cu deposit (Almería, SE Spain)
-
- Journal:
- Clays and Clay Minerals / Volume 57 / Issue 1 / February 2009
- Published online by Cambridge University Press:
- 01 January 2024, pp. 1-24
-
- Article
- Export citation
Forthcoming Papers
-
- Journal:
- Clays and Clay Minerals / Volume 52 / Issue 1 / February 2004
- Published online by Cambridge University Press:
- 01 January 2024, p. 141
-
- Article
-
- You have access
- Export citation
An Endless Capacity for Dissembling: Representing Teenage Girls on the American Stage from The Children's Hour through If Pretty Hurts Ugly Must Be a Muhfucka
-
- Journal:
- Theatre Survey / Volume 65 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 18 March 2024, pp. 37-59
- Print publication:
- January 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The Sharing of the Profits of the Carrera de Indias: The Actors of the Hispanic Colonial Trade and Their Monopolistic Practices in the Second Half of the Eighteenth Century
-
- Journal:
- The Americas / Volume 81 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 09 January 2024, pp. 39-66
- Print publication:
- January 2024
-
- Article
- Export citation
ARTWORK: ED COOPER
-
- Article
- Export citation
Sorption and Direct Electrochemistry of Mitochondrial Cytochrome C on Hematite Surfaces
-
- Journal:
- Clays and Clay Minerals / Volume 53 / Issue 6 / December 2005
- Published online by Cambridge University Press:
- 01 January 2024, pp. 564-571
-
- Article
- Export citation
Powder X-Ray Diffraction Study of the Hydration and Leaching Behavior of Nontronite
-
- Journal:
- Clays and Clay Minerals / Volume 59 / Issue 6 / December 2011
- Published online by Cambridge University Press:
- 01 January 2024, pp. 560-567
-
- Article
- Export citation
Origin of Saponite-Rich Clays in A Fossil Serpentinite-Hosted Hydrothermal System in The Crustal Basement of The Hyblean Plateau (Sicily, Italy)
-
- Journal:
- Clays and Clay Minerals / Volume 60 / Issue 1 / February 2012
- Published online by Cambridge University Press:
- 01 January 2024, pp. 18-31
-
- Article
- Export citation
Measurement of clay surface areas by polyvinylpyrrolidone (PVP) sorption and its use for quantifying illite and smectite abundance
-
- Journal:
- Clays and Clay Minerals / Volume 52 / Issue 5 / October 2004
- Published online by Cambridge University Press:
- 01 January 2024, pp. 589-602
-
- Article
- Export citation
AJL volume 14 issue 1 Cover and Front matter
-
- Journal:
- Asian Journal of International Law / Volume 14 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 23 January 2024, pp. f1-f7
- Print publication:
- January 2024
-
- Article
-
- You have access
- Export citation
