Refine search
Actions for selected content:
238502 results in Physics and Astronomy
3 - Continuum Theory of Electromagnetism and Gravity
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 110-176
-
- Chapter
- Export citation
Fifty years of astrobiology: mapping researcher communities with topic-based network analyses
-
- Journal:
- International Journal of Astrobiology / Volume 24 / 2025
- Published online by Cambridge University Press:
- 13 February 2025, e2
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 1-2
-
- Chapter
- Export citation
Index
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 674-678
-
- Chapter
- Export citation
7 - Nonequilibrium Diffusive Transport
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 393-456
-
- Chapter
- Export citation
Contents
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp v-viii
-
- Chapter
- Export citation
2 - Continuum Mechanics
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 60-109
-
- Chapter
- Export citation
Verifying the Australian MWA EoR pipeline II: Fundamental limits of the AusEoRPipe and the impact of instrumental effects
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 42 / 2025
- Published online by Cambridge University Press:
- 13 February 2025, e024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
5 - Fluid Dynamics
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 261-312
-
- Chapter
- Export citation
The potential of Deception Island, Antarctica, as a multifunctional Martian analogue of astrobiological interest
-
- Journal:
- International Journal of Astrobiology / Volume 24 / 2025
- Published online by Cambridge University Press:
- 13 February 2025, e3
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
11 - Contour-Integration Methods and Applications
- from Part II - Mathematical Methods
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 570-626
-
- Chapter
- Export citation
4 - Elasticity and Elastodynamics
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 177-260
-
- Chapter
- Export citation
1 - An Introduction to Tensor Calculus
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 3-59
-
- Chapter
- Export citation
References
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 672-673
-
- Chapter
- Export citation
Preface
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp ix-xiv
-
- Chapter
- Export citation
6 - Equilibrium Thermodynamics
- from Part I - Continuum Physics
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 313-392
-
- Chapter
- Export citation
9 - The Fourier Series
- from Part II - Mathematical Methods
-
- Book:
- An Introduction to Continuum Physics
- Published online:
- 06 February 2025
- Print publication:
- 13 February 2025, pp 489-518
-
- Chapter
- Export citation
Altitude estimation of radio frequency interference sources via interferometric near-field corrections
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 42 / 2025
- Published online by Cambridge University Press:
- 12 February 2025, e010
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Radio-frequency interference (RFI) presents a significant obstacle to current radio interferometry experiments aimed at the Epoch of Reionization. RFI contamination is often several orders of magnitude brighter than the astrophysical signals of interest, necessitating highly precise identification and flagging. Although existing RFI flagging tools have achieved some success, the pervasive nature of this contamination leads to the rejection of excessive data volumes. In this work, we present a way to estimate an RFI emitter’s altitude using near-field corrections. Being able to obtain the precise location of such an emitter could shift the strategy from merely flagging to subtracting or peeling the RFI, allowing us to preserve a higher fraction of usable data. We conduct a preliminary study using a two-minute observation from the Murchison-Widefield Array (MWA) in which an unknown object briefly crosses the field of view, reflecting RFI signals into the array. By applying near-field corrections that bring the object into focus, we are able to estimate its approximate altitude and speed to be
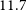 $11.7$ km and 792 km/h, respectively. This allows us to confidently conclude that the object in question is in fact an airplane. We further validate our technique through the analysis of two additional RFI-containing MWA observations, where we are consistently able to identify airplanes as the source of the interference.
$11.7$ km and 792 km/h, respectively. This allows us to confidently conclude that the object in question is in fact an airplane. We further validate our technique through the analysis of two additional RFI-containing MWA observations, where we are consistently able to identify airplanes as the source of the interference.
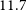
ASKAP EMU radio detection of the reflection Nebula VdB-80 in the Monoceros crossbones filamentary structure
-
- Journal:
- Publications of the Astronomical Society of Australia / Volume 42 / 2025
- Published online by Cambridge University Press:
- 12 February 2025, e032
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
We present a new radio detection from the Australian Square Kilometre Array Pathfinder Evolutionary Map of the Universe (EMU) survey associated with the Reflection Nebula (RN) VdB-80. The radio detection is determined to be a previously unidentified Hii region, now named Lagotis. The RN is located towards Monoceros, centred in the molecular cloud feature known as the ‘Crossbones’. The 944 MHz EMU image shows a roughly semicircular Hii region with an integrated flux density of 30.2
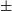 $\pm$0.3 mJy. The Hii region is also seen at 1.4 GHz by NRAO VLA Sky Survey (NVSS), yielding an estimated spectral index of 0.65
$\pm$0.3 mJy. The Hii region is also seen at 1.4 GHz by NRAO VLA Sky Survey (NVSS), yielding an estimated spectral index of 0.65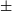 $\pm$0.51, consistent with thermal radio emission. Gaia Data Release 3 (DR3) and Two Micron All Sky Survey (2MASS) data give a distance to the stars associated with the Hii region of
$\pm$0.51, consistent with thermal radio emission. Gaia Data Release 3 (DR3) and Two Micron All Sky Survey (2MASS) data give a distance to the stars associated with the Hii region of 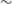 $\sim$960 pc. This implies a size of 0.76
$\sim$960 pc. This implies a size of 0.76 $\times$0.68(
$\times$0.68(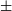 $\pm$0.09) pc for the Hii region. We derive an Hii region electron density of the bright radio feature to be 26 cm
$\pm$0.09) pc for the Hii region. We derive an Hii region electron density of the bright radio feature to be 26 cm $^{-3}$, requiring a Lyman-alpha photon flux of
$^{-3}$, requiring a Lyman-alpha photon flux of 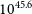 $10^{45.6}$ s
$10^{45.6}$ s $^{-1}$, which is consistent with the expected Lyman flux of HD 46060, the B2 ii type star which is the likely ionising star of the region. The derived distance to this region implies that the Crossbones feature is a superposition of two filamentary clouds, with Lagotis embedded in the far cloud.
$^{-1}$, which is consistent with the expected Lyman flux of HD 46060, the B2 ii type star which is the likely ionising star of the region. The derived distance to this region implies that the Crossbones feature is a superposition of two filamentary clouds, with Lagotis embedded in the far cloud.
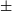
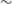

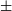

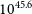

Interaction of a counter-propagating relativistic laser pair with subwavelength thin solid-density foil
-
- Journal:
- High Power Laser Science and Engineering / Volume 13 / 2025
- Published online by Cambridge University Press:
- 12 February 2025, e9
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
