Refine listing
Actions for selected content:
27 results in 37Mxx
Ageing and mortality of persons with HIV: a novel data-driven approach
- Part of
-
- Journal:
- European Journal of Applied Mathematics , First View
- Published online by Cambridge University Press:
- 28 November 2025, pp. 1-27
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
RESERVOIR TIME SERIES ANALYSIS: DEVELOPING RESERVOIR COMPUTING TECHNIQUES FOR NONLINEAR TIME SERIES ANALYSIS FROM FOUNDATIONS TO APPLICATION
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 112 / Issue 2 / October 2025
- Published online by Cambridge University Press:
- 28 July 2025, pp. 397-398
- Print publication:
- October 2025
-
- Article
-
- You have access
- HTML
- Export citation
BALANCING STRUCTURE AND DYNAMICS: DYNAMICAL NETWORKS AND EMBEDDING AS A MODELLING PARADIGM
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 112 / Issue 3 / December 2025
- Published online by Cambridge University Press:
- 04 July 2025, pp. 570-571
- Print publication:
- December 2025
-
- Article
-
- You have access
- HTML
- Export citation
Approximation of the invariant measure for stable stochastic differential equations by the Euler–Maruyama scheme with decreasing step sizes
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 57 / Issue 3 / September 2025
- Published online by Cambridge University Press:
- 31 March 2025, pp. 1000-1030
- Print publication:
- September 2025
-
- Article
- Export citation
-
Let
 $(X_t)_{t \geqslant 0}$ be the solution of the stochastic differential equation where
$(X_t)_{t \geqslant 0}$ be the solution of the stochastic differential equation where \[\mathrm{d} X_t = b(X_t) \,\mathrm{d} t+A\,\mathrm{d} Z_t, \quad X_{0}=x,\]
\[\mathrm{d} X_t = b(X_t) \,\mathrm{d} t+A\,\mathrm{d} Z_t, \quad X_{0}=x,\] $b\,:\, \mathbb{R}^d \rightarrow \mathbb{R}^d$ is a Lipschitz-continuous function,
$b\,:\, \mathbb{R}^d \rightarrow \mathbb{R}^d$ is a Lipschitz-continuous function,  $A \in \mathbb{R}^{d \times d}$ is a positive-definite matrix,
$A \in \mathbb{R}^{d \times d}$ is a positive-definite matrix,  $(Z_t)_{t\geqslant 0}$ is a d-dimensional rotationally symmetric
$(Z_t)_{t\geqslant 0}$ is a d-dimensional rotationally symmetric  $\alpha$-stable Lévy process with
$\alpha$-stable Lévy process with  $\alpha \in (1,2)$ and
$\alpha \in (1,2)$ and  $x\in\mathbb{R}^{d}$. We use two Euler–Maruyama schemes with decreasing step sizes
$x\in\mathbb{R}^{d}$. We use two Euler–Maruyama schemes with decreasing step sizes  $\Gamma = (\gamma_n)_{n\in \mathbb{N}}$ to approximate the invariant measure of
$\Gamma = (\gamma_n)_{n\in \mathbb{N}}$ to approximate the invariant measure of  $(X_t)_{t \geqslant 0}$: one uses independent and identically distributed
$(X_t)_{t \geqslant 0}$: one uses independent and identically distributed  $\alpha$-stable random variables as innovations, and the other employs independent and identically distributed Pareto random variables. We study the convergence rates of these two approximation schemes in the Wasserstein-1 distance. For the first scheme, under the assumption that the function b is Lipschitz and satisfies a certain dissipation condition, we demonstrate a convergence rate of
$\alpha$-stable random variables as innovations, and the other employs independent and identically distributed Pareto random variables. We study the convergence rates of these two approximation schemes in the Wasserstein-1 distance. For the first scheme, under the assumption that the function b is Lipschitz and satisfies a certain dissipation condition, we demonstrate a convergence rate of  $\gamma^{\frac{1}{\alpha}}_n$. This convergence rate can be improved to
$\gamma^{\frac{1}{\alpha}}_n$. This convergence rate can be improved to  $\gamma^{1+\frac {1}{\alpha}-\frac{1}{\kappa}}_n$ for any
$\gamma^{1+\frac {1}{\alpha}-\frac{1}{\kappa}}_n$ for any  $\kappa \in [1,\alpha)$, provided b has the additional regularity of bounded second-order directional derivatives. For the second scheme, where the function b is assumed to be twice continuously differentiable, we establish a convergence rate of
$\kappa \in [1,\alpha)$, provided b has the additional regularity of bounded second-order directional derivatives. For the second scheme, where the function b is assumed to be twice continuously differentiable, we establish a convergence rate of  $\gamma^{\frac{2-\alpha}{\alpha}}_n$; moreover, we show that this rate is optimal for the one-dimensional stable Ornstein–Uhlenbeck process. Our theorems indicate that the recent significant result of [34] concerning the unadjusted Langevin algorithm with additive innovations can be extended to stochastic differential equations driven by an
$\gamma^{\frac{2-\alpha}{\alpha}}_n$; moreover, we show that this rate is optimal for the one-dimensional stable Ornstein–Uhlenbeck process. Our theorems indicate that the recent significant result of [34] concerning the unadjusted Langevin algorithm with additive innovations can be extended to stochastic differential equations driven by an  $\alpha$-stable Lévy process and that the corresponding convergence rate exhibits similar behaviour. Compared with the result in [6], our assumptions have relaxed the second-order differentiability condition, requiring only a Lipschitz condition for the first scheme, which broadens the applicability of our approach.
$\alpha$-stable Lévy process and that the corresponding convergence rate exhibits similar behaviour. Compared with the result in [6], our assumptions have relaxed the second-order differentiability condition, requiring only a Lipschitz condition for the first scheme, which broadens the applicability of our approach.
















On the Ziv–Merhav theorem beyond Markovianity I
- Part of
-
- Journal:
- Canadian Journal of Mathematics / Volume 77 / Issue 3 / June 2025
- Published online by Cambridge University Press:
- 07 March 2024, pp. 891-915
- Print publication:
- June 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
BURSTING SOLUTIONS OF THE RÖSSLER EQUATIONS
- Part of
-
- Journal:
- The ANZIAM Journal / Volume 65 / Issue 1-2 / January 2023
- Published online by Cambridge University Press:
- 10 August 2023, pp. 93-110
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
We provide an analytic solution of the Rössler equations based on the asymptotic limit
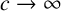 $c\to \infty $ and we show in this limit that the solution takes the form of multiple pulses, similar to “burst” firing of neurons. We are able to derive an approximate Poincaré map for the solutions, which compares reasonably with a numerically derived map.
$c\to \infty $ and we show in this limit that the solution takes the form of multiple pulses, similar to “burst” firing of neurons. We are able to derive an approximate Poincaré map for the solutions, which compares reasonably with a numerically derived map.
The aromatic bicomplex for the description of divergence-free aromatic forms and volume-preserving integrators
- Part of
-
- Journal:
- Forum of Mathematics, Sigma / Volume 11 / 2023
- Published online by Cambridge University Press:
- 08 August 2023, e69
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Overcoming the timescale barrier in molecular dynamics: Transfer operators, variational principles and machine learning
- Part of
-
- Journal:
- Acta Numerica / Volume 32 / May 2023
- Published online by Cambridge University Press:
- 11 May 2023, pp. 517-673
-
- Article
-
- You have access
- Open access
- Export citation
A QUANTITATIVE EVALUATION OF LAGRANGIAN COHERENT STRUCTURE DETECTION METHODS BASED ON COMPUTATIONAL AND EXPERIMENTAL LIMITATIONS
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 107 / Issue 1 / February 2023
- Published online by Cambridge University Press:
- 05 December 2022, pp. 173-174
- Print publication:
- February 2023
-
- Article
-
- You have access
- HTML
- Export citation
STATISTICAL STABILITY FOR DETERMINISTIC AND RANDOM DYNAMICAL SYSTEMS
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 105 / Issue 1 / February 2022
- Published online by Cambridge University Press:
- 30 September 2021, pp. 171-172
- Print publication:
- February 2022
-
- Article
-
- You have access
- HTML
- Export citation
Deep learning models for global coordinate transformations that linearise PDEs
- Part of
-
- Journal:
- European Journal of Applied Mathematics / Volume 32 / Issue 3 / June 2021
- Published online by Cambridge University Press:
- 24 September 2020, pp. 515-539
-
- Article
- Export citation
Approximating integrals with respect to stationary probability measures of iterated function systems
- Part of
-
- Journal:
- Ergodic Theory and Dynamical Systems / Volume 41 / Issue 8 / August 2021
- Published online by Cambridge University Press:
- 08 June 2020, pp. 2294-2310
- Print publication:
- August 2021
-
- Article
- Export citation
NONLINEAR TIME SERIES ANALYSIS USING ORDINAL NETWORKS WITH SELECT APPLICATIONS IN BIOMEDICAL SIGNAL PROCESSING
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 100 / Issue 1 / August 2019
- Published online by Cambridge University Press:
- 17 May 2019, pp. 170-172
- Print publication:
- August 2019
-
- Article
-
- You have access
- Export citation
COUPLE MICROSCALE PERIODIC PATCHES TO SIMULATE MACROSCALE EMERGENT DYNAMICS
- Part of
-
- Journal:
- The ANZIAM Journal / Volume 59 / Issue 3 / January 2018
- Published online by Cambridge University Press:
- 30 January 2018, pp. 313-334
-
- Article
-
- You have access
- Export citation
Novel Conformal Structure-Preserving Algorithms for Coupled Damped Nonlinear Schrödinger System
- Part of
-
- Journal:
- Advances in Applied Mathematics and Mechanics / Volume 9 / Issue 6 / December 2017
- Published online by Cambridge University Press:
- 28 November 2017, pp. 1383-1403
- Print publication:
- December 2017
-
- Article
- Export citation
Adaptive Stokes Preconditioning for Steady Incompressible Flows
- Part of
- Numerical problems in dynamical systems
- Equations of mathematical physics and other areas of application
- Approximation methods and numerical treatment of dynamical systems
- Applications - Dynamical systems and ergodic theory
- Numerical linear algebra
- Incompressible viscous fluids
- Hydrodynamic stability
-
- Journal:
- Communications in Computational Physics / Volume 22 / Issue 2 / August 2017
- Published online by Cambridge University Press:
- 21 June 2017, pp. 494-516
- Print publication:
- August 2017
-
- Article
- Export citation
-
This paper describes an adaptive preconditioner for numerical continuation of incompressible Navier–Stokes flows based on Stokes preconditioning [42] which has been used successfully in studies of pattern formation in convection. The preconditioner takes the form of the Helmholtz operator I–ΔtL which maps the identity (no preconditioner) for Δt≪1 to Laplacian preconditioning for Δt≫1. It is built on a first order Euler time-discretization scheme and is part of the family of matrix-free methods. The preconditioner is tested on two fluid configurations: three-dimensional doubly diffusive convection and a two-dimensional projection of a shear flow. In the former case, it is found that Stokes preconditioning is more efficient for
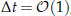 , away from the values used in the literature. In the latter case, the simple use of the preconditioner is not sufficient and it is necessary to split the system of equations into two subsystems which are solved simultaneously using two different preconditioners, one of which is parameter dependent. Due to the nature of these applications and the flexibility of the approach described, this preconditioner is expected to help in a wide range of applications.
, away from the values used in the literature. In the latter case, the simple use of the preconditioner is not sufficient and it is necessary to split the system of equations into two subsystems which are solved simultaneously using two different preconditioners, one of which is parameter dependent. Due to the nature of these applications and the flexibility of the approach described, this preconditioner is expected to help in a wide range of applications.
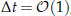 , away from the values used in the literature. In the latter case, the simple use of the preconditioner is not sufficient and it is necessary to split the system of equations into two subsystems which are solved simultaneously using two different preconditioners, one of which is parameter dependent. Due to the nature of these applications and the flexibility of the approach described, this preconditioner is expected to help in a wide range of applications.
, away from the values used in the literature. In the latter case, the simple use of the preconditioner is not sufficient and it is necessary to split the system of equations into two subsystems which are solved simultaneously using two different preconditioners, one of which is parameter dependent. Due to the nature of these applications and the flexibility of the approach described, this preconditioner is expected to help in a wide range of applications.The Landau-Zener Transition and the Surface Hopping Method for the 2D Dirac Equation for Graphene
- Part of
- Applications to specific physical systems
- General mathematical topics and methods in quantum theory
- Partial differential equations, initial value and time-dependent initial-boundary value problems
- Quantum theory
- Equations of mathematical physics and other areas of application
- Approximation methods and numerical treatment of dynamical systems
-
- Journal:
- Communications in Computational Physics / Volume 21 / Issue 2 / February 2017
- Published online by Cambridge University Press:
- 07 February 2017, pp. 313-357
- Print publication:
- February 2017
-
- Article
- Export citation
ON ASPECTS OF NUMERICAL ERGODIC THEORY: STABILITY OF ULAM’S METHOD, COMPUTING OSELEDETS SUBSPACES AND OPTIMAL MIXING
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 95 / Issue 1 / February 2017
- Published online by Cambridge University Press:
- 23 November 2016, pp. 165-166
- Print publication:
- February 2017
-
- Article
-
- You have access
- Export citation
Scale Transitions in Magnetisation Dynamics
- Part of
-
- Journal:
- Communications in Computational Physics / Volume 20 / Issue 4 / October 2016
- Published online by Cambridge University Press:
- 05 October 2016, pp. 969-988
- Print publication:
- October 2016
-
- Article
- Export citation
EXPECTATIONS OVER DETERMINISTIC FRACTAL SETS
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 94 / Issue 1 / August 2016
- Published online by Cambridge University Press:
- 25 April 2016, pp. 171-172
- Print publication:
- August 2016
-
- Article
-
- You have access
- Export citation
