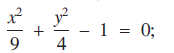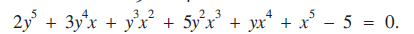Refine listing
Actions for selected content:
1418670 results in Open Access
CNJ volume 68 issue 4 Cover and Back matter
-
- Journal:
- Canadian Journal of Linguistics/Revue canadienne de linguistique / Volume 68 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 15 February 2024, pp. b1-b5
-
- Article
-
- You have access
- Export citation
Kierkegaard and Schmitt on the State of Exception
-
- Journal:
- Journal of Law and Religion / Volume 39 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 1-15
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
108.05 Ramanujan’s proof of Bertrand’s postulate
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 130-134
- Print publication:
- March 2024
-
- Article
- Export citation
108.15 The Madhava-Leibniz theorem
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 149-151
- Print publication:
- March 2024
-
- Article
- Export citation
Sufficient conditions for non-zero entropy of closed relations
- Part of
-
- Journal:
- Ergodic Theory and Dynamical Systems / Volume 44 / Issue 11 / November 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 3091-3119
- Print publication:
- November 2024
-
- Article
- Export citation
ABC-triangles
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 78-83
- Print publication:
- March 2024
-
- Article
- Export citation
108.10 Proof without words:
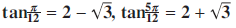 ${\rm{tan }}{\pi\over {{\rm{12}}}}{\rm{=2 - }}\sqrt 3 {\rm{, tan }}{{{\rm{5}}\pi } \over {{\rm{12}}}}{\rm{=2 + }}\sqrt {\rm{3}} $
${\rm{tan }}{\pi\over {{\rm{12}}}}{\rm{=2 - }}\sqrt 3 {\rm{, tan }}{{{\rm{5}}\pi } \over {{\rm{12}}}}{\rm{=2 + }}\sqrt {\rm{3}} $
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 143-144
- Print publication:
- March 2024
-
- Article
- Export citation
108.08 Cone and Integral
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 140-142
- Print publication:
- March 2024
-
- Article
- Export citation
Walk on a grid
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 111-117
- Print publication:
- March 2024
-
- Article
- Export citation
Extensions of Vittas’ Theorem
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 53-60
- Print publication:
- March 2024
-
- Article
- Export citation
Axé as the cornerstone of Candomblé philosophy and its significance for an understanding of well-being (bem estar)
-
- Journal:
- Religious Studies / Volume 61 / Issue 2 / June 2025
- Published online by Cambridge University Press:
- 15 February 2024, pp. 453-465
- Print publication:
- June 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Who, Why, When, and Where From? The Peopling of the Canary Islands and the Challenges of Archaeometry
-
- Journal:
- European Journal of Archaeology / Volume 27 / Issue 2 / May 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 192-209
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
108.16 Golden triangles founded on Kepler’s triangle
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 152-154
- Print publication:
- March 2024
-
- Article
- Export citation
Irrationality and transcendence in number theory by David Angell , pp. 242, £59.99, (hard), ISBN 978-0-367-62837-6, Chapman and Hall/CRC (2022)
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 180-182
- Print publication:
- March 2024
-
- Article
- Export citation
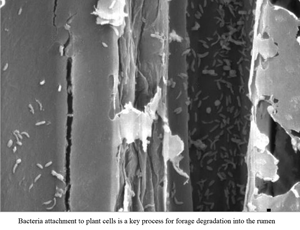
Feed intake, microbial adherence and fibrolytic activity in residues of forage samples incubated in the rumen of sheep fed grass forages and/or a total mixed ration
-
- Journal:
- The Journal of Agricultural Science / Volume 161 / Issue 6 / December 2023
- Published online by Cambridge University Press:
- 15 February 2024, pp. 871-876
-
- Article
- Export citation
The Devil’s Advocate and the binomial expansion
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 161-162
- Print publication:
- March 2024
-
- Article
- Export citation
Geoffrey Lloyd and Aparecida Vilaça, Of Jaguars and Butterflies: Metalogues on Issues in Anthropology and Philosophy Oxford: Berghahn Books, 2023. Pp. 150. ISBN 978-1-80073-904-8. £89.00 (hardback).
-
- Journal:
- The British Journal for the History of Science / Volume 57 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 312-314
- Print publication:
- June 2024
-
- Article
- Export citation
MAG volume 108 issue 571 Cover and Front matter
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. f1-f2
- Print publication:
- March 2024
-
- Article
-
- You have access
- Export citation
COINTEGRATING POLYNOMIAL REGRESSIONS: ROBUSTNESS OF FULLY MODIFIED OLS
-
- Journal:
- Econometric Theory / Volume 41 / Issue 3 / June 2025
- Published online by Cambridge University Press:
- 15 February 2024, pp. 688-708
-
- Article
-
- You have access
- Open access
- Export citation
xy = cos (x + y) and other implicit equations that are surprisingly easy to plot
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 1-11
- Print publication:
- March 2024
-
- Article
- Export citation
-
The following equations relate y only implicitly to x:(1)
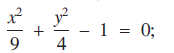 (2)
(2)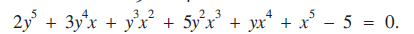 In both equations, y is a function of x for a continuous range of (x, y) values in the real x-y plane. (1) represents an ellipse. (2) has been designed by the author to have a solution in the real x-y plane at (−1, 2), and because the function on the left-hand side of (2) meets certain conditions regarding continuity and partial differentiability there must be a line of points in the real x-y plane satisfying (2) and passing continuously through (−1, 2) [1, pp. 23-28].
In both equations, y is a function of x for a continuous range of (x, y) values in the real x-y plane. (1) represents an ellipse. (2) has been designed by the author to have a solution in the real x-y plane at (−1, 2), and because the function on the left-hand side of (2) meets certain conditions regarding continuity and partial differentiability there must be a line of points in the real x-y plane satisfying (2) and passing continuously through (−1, 2) [1, pp. 23-28].