Refine listing
Actions for selected content:
1418667 results in Open Access
Post-growth peacebuilding
-
- Journal:
- Review of International Studies / Volume 50 / Issue 5 / September 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 877-887
- Print publication:
- September 2024
-
- Article
- Export citation
Theory of infinite sequences and series by Ludmila Bourchteinand Andrei Bourchtein, pp. 377, £54.99, (paper), ISBN 978-3-03079-430-9, Springer Verlag (2022)
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 182-183
- Print publication:
- March 2024
-
- Article
- Export citation
GRAPH CHARACTERISATION OF THE ANNIHILATOR IDEALS OF LEAVITT PATH ALGEBRAS
- Part of
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 110 / Issue 3 / December 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 498-507
- Print publication:
- December 2024
-
- Article
- Export citation
-
If E is a graph and K is a field, we consider an ideal I of the Leavitt path algebra
 $L_K(E)$ of E over K. We describe the admissible pair corresponding to the smallest graded ideal which contains I where the grading in question is the natural grading of
$L_K(E)$ of E over K. We describe the admissible pair corresponding to the smallest graded ideal which contains I where the grading in question is the natural grading of  $L_K(E)$ by
$L_K(E)$ by  ${\mathbb {Z}}$. Using this description, we show that the right and the left annihilators of I are equal (which may be somewhat surprising given that I may not be self-adjoint). In particular, we establish that both annihilators correspond to the same admissible pair and its description produces the characterisation from the title. Then, we turn to the property that the right (equivalently left) annihilator of any ideal is a direct summand and recall that a unital ring with this property is said to be quasi-Baer. We exhibit a condition on E which is equivalent to unital
${\mathbb {Z}}$. Using this description, we show that the right and the left annihilators of I are equal (which may be somewhat surprising given that I may not be self-adjoint). In particular, we establish that both annihilators correspond to the same admissible pair and its description produces the characterisation from the title. Then, we turn to the property that the right (equivalently left) annihilator of any ideal is a direct summand and recall that a unital ring with this property is said to be quasi-Baer. We exhibit a condition on E which is equivalent to unital  $L_K(E)$ having this property.
$L_K(E)$ having this property.




Patterns among square roots of the 2 × 2 identity matrix
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 84-93
- Print publication:
- March 2024
-
- Article
- Export citation
-
The 2 × 2 identity matrix,
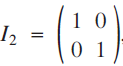 $${I_2} = \left( \begin{gathered}{\rm{1 \,\,\,0}} \hfill \\{\rm{0 \,\,\,1}} \hfill \\ \end{gathered}\right)$$, has an infinite number of square roots. The purpose of this paper is to show some interesting patterns that appear among these square roots. In the process, we will take a brief tour of some topics in number theory, including Pythagorean triples, Eisenstein triples, Fibonacci numbers, Pell numbers and Diophantine triples.
$${I_2} = \left( \begin{gathered}{\rm{1 \,\,\,0}} \hfill \\{\rm{0 \,\,\,1}} \hfill \\ \end{gathered}\right)$$, has an infinite number of square roots. The purpose of this paper is to show some interesting patterns that appear among these square roots. In the process, we will take a brief tour of some topics in number theory, including Pythagorean triples, Eisenstein triples, Fibonacci numbers, Pell numbers and Diophantine triples.
CNJ volume 68 issue 4 Cover and Front matter
-
- Journal:
- Canadian Journal of Linguistics/Revue canadienne de linguistique / Volume 68 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 15 February 2024, pp. f1-f2
-
- Article
-
- You have access
- Export citation
Non-linear relationship between the body roundness index and metabolic syndrome: data from National Health and Nutrition Examination Survey (NHANES) 1999–2018
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 11 / 14 June 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 1852-1859
- Print publication:
- 14 June 2024
-
- Article
-
- You have access
- HTML
- Export citation
Thank You to Our Reviewers
-
- Journal:
- Nationalities Papers / Volume 52 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 233-235
-
- Article
- Export citation
TRILINEAR FOURIER MULTIPLIERS ON HARDY SPACES
- Part of
-
- Journal:
- Journal of the Institute of Mathematics of Jussieu / Volume 23 / Issue 5 / September 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 2217-2278
- Print publication:
- September 2024
-
- Article
- Export citation
-
In this paper, we obtain the
 $H^{p_1}\times H^{p_2}\times H^{p_3}\to H^p$ boundedness for trilinear Fourier multiplier operators, which is a trilinear analogue of the multiplier theorem of Calderón and Torchinsky [4]. Our result improves the trilinear estimate in [22] by additionally assuming an appropriate vanishing moment condition, which is natural in the boundedness into the Hardy space
$H^{p_1}\times H^{p_2}\times H^{p_3}\to H^p$ boundedness for trilinear Fourier multiplier operators, which is a trilinear analogue of the multiplier theorem of Calderón and Torchinsky [4]. Our result improves the trilinear estimate in [22] by additionally assuming an appropriate vanishing moment condition, which is natural in the boundedness into the Hardy space 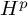 $H^p$ for
$H^p$ for 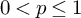 $0<p\le 1$.
$0<p\le 1$.

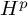
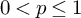
Turning expletive: From embedded speech-acts to embedded propositions
-
- Journal:
- Canadian Journal of Linguistics/Revue canadienne de linguistique / Volume 68 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 15 February 2024, pp. 590-614
-
- Article
-
- You have access
- HTML
- Export citation
Christopher D.E. Willoughby, Masters of Health: Racial Science and Slavery in U.S. Medical Schools Chapel Hill: University of North Carolina Press, 2022. Pp. 282. ISBN 978-1-469-67184-0. $99.00 (hardcover).
-
- Journal:
- The British Journal for the History of Science / Volume 57 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 492-493
- Print publication:
- September 2024
-
- Article
- Export citation
108.17 On a generalisation of the Lemoine axis
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 154-158
- Print publication:
- March 2024
-
- Article
- Export citation
108.13 Indeterminate exponentials without tears
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 146-148
- Print publication:
- March 2024
-
- Article
- Export citation
Railways’ Economic Impact on Uttar Pradesh and Colonial North India (1860–1914): The Iron Raj By Ian D. Derbyshire (review). 615 pp. Newcastle-upon-Tyne, Cambridge Scholars Publishing, 2022.
-
- Journal:
- Journal of the Royal Asiatic Society / Volume 34 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 484-485
- Print publication:
- April 2024
-
- Article
- Export citation
Rearing and 60Co radiation do not affect attractiveness but alter the volatile profiles released by Anastrepha obliqua calling males
-
- Journal:
- Bulletin of Entomological Research / Volume 114 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 237-243
-
- Article
- Export citation
108.12 Proof without words: a lower bound for n!
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, p. 146
- Print publication:
- March 2024
-
- Article
- Export citation
Evolving An African Postcolonial Condition: Cultural Property Restitution, Cinematic Independence, Globalized NGO Compassion, and Grappling with an Elite Corruption Complex
-
- Journal:
- African Studies Review / Volume 67 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 431-441
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Michael C. Rea Essays in Analytic Theology 2 vols (Oxford: Oxford University Press, 2021). Pp. 576. $150.00 (Hbk). ISBN 9780198866794.
-
- Journal:
- Religious Studies / Volume 61 / Issue 2 / June 2025
- Published online by Cambridge University Press:
- 15 February 2024, pp. 552-554
- Print publication:
- June 2025
-
- Article
- Export citation
Gender, Politics, and (Missing) Data: Evidence from the Pacific Island Countries and Territories
-
- Journal:
- Politics & Gender / Volume 20 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 246-251
-
- Article
-
- You have access
- HTML
- Export citation
Change and variations, a history of differential equations to 1900 by Jeremy Gray , pp. 261, £29.99, (hard), ISBN 978-3-03070-574-9, Springer Verlag (2021)
-
- Journal:
- The Mathematical Gazette / Volume 108 / Issue 571 / March 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 176-177
- Print publication:
- March 2024
-
- Article
- Export citation
Integration of traditional and telematics data for efficient insurance claims prediction
-
- Journal:
- ASTIN Bulletin: The Journal of the IAA / Volume 54 / Issue 2 / May 2024
- Published online by Cambridge University Press:
- 15 February 2024, pp. 263-279
- Print publication:
- May 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
