Refine listing
Actions for selected content:
1418740 results in Open Access
Antibiotic consumption in French nursing homes between 2018 and 2022: A multicenter survey
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 45 / Issue 6 / June 2024
- Published online by Cambridge University Press:
- 19 February 2024, pp. 740-745
- Print publication:
- June 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Constructions of Racial Savagery in Early Twentieth-Century US Narratives of White Civilization
-
- Journal:
- Journal of American Studies / Volume 58 / Issue 2 / May 2024
- Published online by Cambridge University Press:
- 19 February 2024, pp. 193-219
- Print publication:
- May 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
‘Best paper’ prize winners for 2023 and in the current issue: entry to UK ENT specialist training
-
- Journal:
- The Journal of Laryngology & Otology / Volume 138 / Issue 3 / March 2024
- Published online by Cambridge University Press:
- 16 February 2024, p. 237
- Print publication:
- March 2024
-
- Article
-
- You have access
- HTML
- Export citation
PERFECTING GROUP SCHEMES
- Part of
-
- Journal:
- Journal of the Institute of Mathematics of Jussieu / Volume 23 / Issue 6 / November 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 2549-2591
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Academic Solidarity in the Wake of Disaster: Blueprint for an Online Writing Support Group
-
- Journal:
- PS: Political Science & Politics / Volume 57 / Issue 3 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 370-377
- Print publication:
- July 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Comparative Studies of Russian and European Welfare Polities – CORRIGENDUM
-
- Journal:
- Social Policy and Society / Volume 23 / Issue 3 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, p. 803
- Print publication:
- July 2024
-
- Article
-
- You have access
- HTML
- Export citation
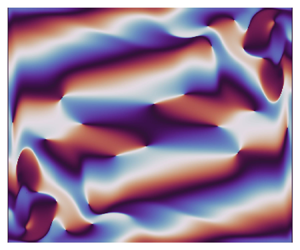
Vertical convection regimes in a two-dimensional rectangular cavity: Prandtl and aspect ratio dependence
-
- Journal:
- Journal of Fluid Mechanics / Volume 981 / 25 February 2024
- Published online by Cambridge University Press:
- 16 February 2024, A10
-
- Article
- Export citation
-
Vertical convection is the fluid motion that is induced by the heating and cooling of two opposed vertical boundaries of a rectangular cavity (see e.g. Wang et al., J. Fluid Mech., vol. 917, 2021, A6). We consider the linear stability of the steady two-dimensional flow reached at Rayleigh numbers of O(
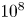 $10^8$). As a function of the Prandtl number,
$10^8$). As a function of the Prandtl number, 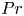 $Pr$, and the height-to-width aspect ratio of the domain,
$Pr$, and the height-to-width aspect ratio of the domain, 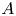 $A$, the base flow of each case is computed numerically and linear simulations are used to obtain the properties of the leading linear instability mode. Flow regimes depend on the presence of a circulation in the entire cavity, detachment of the thermal layer from the boundary or the corner regions and on the oscillation frequency relative to the natural frequency of oscillation in the stably temperature-stratified interior, allowing for the presence of internal waves or not. Accordingly, the regime is called slow or fast, respectively. Either the global circulation or internal waves in the interior may couple the top and bottom buoyancy currents, while their absence implies asymmetry in their perturbation amplitude. Six flow regimes are found in the range of
$A$, the base flow of each case is computed numerically and linear simulations are used to obtain the properties of the leading linear instability mode. Flow regimes depend on the presence of a circulation in the entire cavity, detachment of the thermal layer from the boundary or the corner regions and on the oscillation frequency relative to the natural frequency of oscillation in the stably temperature-stratified interior, allowing for the presence of internal waves or not. Accordingly, the regime is called slow or fast, respectively. Either the global circulation or internal waves in the interior may couple the top and bottom buoyancy currents, while their absence implies asymmetry in their perturbation amplitude. Six flow regimes are found in the range of 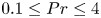 $0.1 \leq Pr \leq 4$ and
$0.1 \leq Pr \leq 4$ and 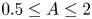 $0.5 \leq A \leq 2$. For
$0.5 \leq A \leq 2$. For 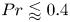 $Pr \lessapprox 0.4$ and
$Pr \lessapprox 0.4$ and 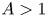 $A>1$, the base flow is driven by a large circulation in the entire cavity. For
$A>1$, the base flow is driven by a large circulation in the entire cavity. For 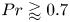 $Pr \gtrapprox 0.7$, the thermal boundary layers are thin and the instability is driven by the motion along the wall and the detached boundary layer. A transition between these regimes is marked by a dramatic change in oscillation frequency at
$Pr \gtrapprox 0.7$, the thermal boundary layers are thin and the instability is driven by the motion along the wall and the detached boundary layer. A transition between these regimes is marked by a dramatic change in oscillation frequency at 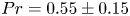 $Pr = 0.55 \pm 0.15$ and
$Pr = 0.55 \pm 0.15$ and 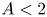 $A <2$.
$A <2$.
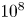
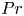
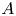
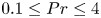
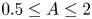
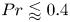
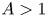
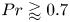
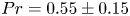
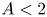
Causal Agnosticism About Race: Variable Selection Problems in Causal Inference
-
- Journal:
- Philosophy of Science / Volume 91 / Issue 5 / December 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 1098-1108
- Print publication:
- December 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Paolo Galluzzi, The Italian Renaissance of Machines Cambridge, MA: Harvard University Press, 2020. Pp. 296. ISBN 978-0-674-98439-4. £37.95 (hardcover).
-
- Journal:
- The British Journal for the History of Science / Volume 57 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 314-316
- Print publication:
- June 2024
-
- Article
- Export citation
Reburials of Eminent Masters: The Construction of Quanzhen Daoist Lineages in North China under Mongol Rule
-
- Journal:
- Journal of Chinese History / Volume 8 / Issue 2 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 297-326
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Rising and settling 2-D cylinders with centre-of-mass offset
-
- Journal:
- Journal of Fluid Mechanics / Volume 981 / 25 February 2024
- Published online by Cambridge University Press:
- 16 February 2024, A7
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The Tang legacy on the Silk Road during the Uighur era: urbanisation in the eastern Tianshan region during the ninth to thirteenth centuries
-
- Journal:
- Journal of the Royal Asiatic Society / Volume 34 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 377-398
- Print publication:
- April 2024
-
- Article
- Export citation
Discursive scaling of solidarity through difference: Experiences of African women in the African diaspora
-
- Journal:
- Language in Society / Volume 54 / Issue 3 / June 2025
- Published online by Cambridge University Press:
- 16 February 2024, pp. 491-513
- Print publication:
- June 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Trilobites from the Al Rose Formation (Lower Ordovician, Inyo Mountains, California)—faunas marginal to the Great Basin
-
- Journal:
- Journal of Paleontology / Volume 98 / Issue 4 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 644-657
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
A single-institutional experience with 36 children less than 5 kilograms supported with the Berlin Heart: Comparison of congenital versus acquired heart disease
-
- Journal:
- Cardiology in the Young / Volume 34 / Issue 6 / June 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 1342-1349
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Effects of the COVID-19 Pandemic on Submissions to Politics & Gender
-
- Journal:
- PS: Political Science & Politics / Volume 57 / Issue 3 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 420-426
- Print publication:
- July 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
By Any Means Necessary? How Black and White Americans Evaluate Protest Tactics in Response to a Police Killing
-
- Journal:
- Journal of Race, Ethnicity and Politics / Volume 9 / Issue 2 / July 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 235-258
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Holocene relative sea-level changes along the Caribbean and Pacific coasts of northwestern South America
-
- Journal:
- Quaternary Research / Volume 119 / May 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 28-43
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
BJN volume 131 issue 6 Cover and Front matter
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 6 / 28 March 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. f1-f2
- Print publication:
- 28 March 2024
-
- Article
-
- You have access
- Export citation
Association between dietary macronutrient composition and plasma one-carbon metabolites and B-vitamin cofactors in patients with stable angina pectoris
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 10 / 28 May 2024
- Published online by Cambridge University Press:
- 16 February 2024, pp. 1678-1690
- Print publication:
- 28 May 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
