Refine listing
Actions for selected content:
1418918 results in Open Access
Growth hegemony and post-growth futures: A complex hegemony approach
-
- Journal:
- Review of International Studies / Volume 50 / Issue 5 / September 2024
- Published online by Cambridge University Press:
- 23 February 2024, pp. 932-942
- Print publication:
- September 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

The modulation of coherent structures by the near-wall motions of particles
-
- Journal:
- Journal of Fluid Mechanics / Volume 981 / 25 February 2024
- Published online by Cambridge University Press:
- 23 February 2024, A26
-
- Article
- Export citation
-
Particle–wall interaction generates strong particle near-wall motion, including collision bounce and impact splashing. To distinguish the effect of particles and particle near-wall motions on the turbulent coherent structure, this study carried out three different cases of sand-laden two-phase flow measurements: a uniform sand release at the top, local-laying sand bed and global-laying sand bed (Liu et al., J. Fluid Mech., vol. 943, 2022, A8). Based on large field of view particle image velocimetry/particle tracking velocimetry measurements, we obtained the velocity field of a two-dimensional gas–solid two-phase dilute faction flow
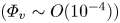 $(\varPhi _{v} \sim O(10^{-4}))$ with a friction Reynolds number
$(\varPhi _{v} \sim O(10^{-4}))$ with a friction Reynolds number 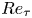 $R e_{\tau }$ of 3950. Results indicate that particles weaken the high- and low-velocity iso-momentum zones and hairpin vortices, resulting in the increased length scale of the coherent structure. However, the collision bounce and impact splashing break up the inner iso-momentum zone and hairpin vortices while enhancing them in the outer region, thus reducing the structure scale. In addition, the upward-moving particles increase the large-scale structure inclination angle, while the downward-moving particles decrease it. The linear coherence spectrum analysis suggests that the particles themselves do not change the structural self-similarity, but their saltation motions disrupt the similarity of the near-wall structure, making the inclination angle decrease with the scale, and the generated ascending particles reduce the aspect ratio of the streamwise to wall-normal direction in the outer region.
$R e_{\tau }$ of 3950. Results indicate that particles weaken the high- and low-velocity iso-momentum zones and hairpin vortices, resulting in the increased length scale of the coherent structure. However, the collision bounce and impact splashing break up the inner iso-momentum zone and hairpin vortices while enhancing them in the outer region, thus reducing the structure scale. In addition, the upward-moving particles increase the large-scale structure inclination angle, while the downward-moving particles decrease it. The linear coherence spectrum analysis suggests that the particles themselves do not change the structural self-similarity, but their saltation motions disrupt the similarity of the near-wall structure, making the inclination angle decrease with the scale, and the generated ascending particles reduce the aspect ratio of the streamwise to wall-normal direction in the outer region.

French historical and contemporary archaeology: a critical assessment
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The effectiveness of cognitive behavioural therapy for depression in women with breast cancer: a systematic review and meta-analysis
-
- Journal:
- Behavioural and Cognitive Psychotherapy / Volume 52 / Issue 4 / July 2024
- Published online by Cambridge University Press:
- 23 February 2024, pp. 394-413
- Print publication:
- July 2024
-
- Article
- Export citation
Bank Competition and Information Production
-
- Journal:
- Journal of Financial and Quantitative Analysis / Volume 59 / Issue 7 / November 2024
- Published online by Cambridge University Press:
- 23 February 2024, pp. 3479-3499
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- Export citation
The role of the peer effect in forming pension expectations among the middle-aged: existence and mechanisms
-
- Journal:
- Journal of Pension Economics & Finance / Volume 23 / Issue 4 / October 2024
- Published online by Cambridge University Press:
- 23 February 2024, pp. 549-573
-
- Article
- Export citation
Temperature fluctuations and ventilation dynamics induced by atmospheric pressure variations in Lamalunga Cave (Apulia, Italy) and their influences on speleothem growth
-
- Journal:
- Quaternary Research / Volume 118 / March 2024
- Published online by Cambridge University Press:
- 23 February 2024, pp. 100-115
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Programmes to manage food selectivity in individuals with autism spectrum disorder
-
- Journal:
- Nutrition Research Reviews / Volume 38 / Issue 1 / June 2025
- Published online by Cambridge University Press:
- 22 February 2024, pp. 112-125
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
NCM volume 20 issue 3 Cover and Back matter
-
- Journal:
- Nineteenth-Century Music Review / Volume 20 / Issue 3 / December 2023
- Published online by Cambridge University Press:
- 22 February 2024, pp. b1-b2
-
- Article
-
- You have access
- Export citation
Variability in phenolic compounds, antioxidant capacity and essential oil profile in different tissues of Ferula assa-foetida L. populations in Iran: an opportunity for industrial products
-
- Journal:
- Plant Genetic Resources / Volume 22 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 22 February 2024, pp. 97-106
-
- Article
- Export citation
The pernicious role of stress on intergenerational continuity of psychopathology
-
- Journal:
- Development and Psychopathology / Volume 36 / Issue 5 / December 2024
- Published online by Cambridge University Press:
- 22 February 2024, pp. 2376-2389
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Higher-order CBT skills: are there differences in meta-competence between trainee and experienced therapists?
-
- Journal:
- The Cognitive Behaviour Therapist / Volume 17 / 2024
- Published online by Cambridge University Press:
- 22 February 2024, e7
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Andrew Kornbluth. The August Trials: The Holocaust and Postwar Justice in Poland Cambridge, MA: Harvard University Press, 2021. Pp. 352.
-
- Journal:
- Austrian History Yearbook / Volume 55 / May 2024
- Published online by Cambridge University Press:
- 22 February 2024, pp. 488-491
- Print publication:
- May 2024
-
- Article
- Export citation
How does trade policy uncertainty affect firms’ pollution emissions? Theory and evidence from China
-
- Journal:
- Macroeconomic Dynamics / Volume 28 / Issue 8 / December 2024
- Published online by Cambridge University Press:
- 22 February 2024, pp. 1776-1808
-
- Article
- Export citation
Music therapy for depression: a Cochrane Review
- Part of
-
- Journal:
- BJPsych Advances / Volume 30 / Issue 2 / March 2024
- Published online by Cambridge University Press:
- 22 February 2024, p. 72
- Print publication:
- March 2024
-
- Article
-
- You have access
- HTML
- Export citation
Radiation and Chemical Program Research for Multi-Utility and Repurposed Countermeasures: A US Department of Health and Human Services Agencies Perspective
-
- Journal:
- Disaster Medicine and Public Health Preparedness / Volume 18 / 2024
- Published online by Cambridge University Press:
- 22 February 2024, e35
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Dietary intake of dicarbonyl compounds and changes in body weight over time in a large cohort of European adults
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 11 / 14 June 2024
- Published online by Cambridge University Press:
- 22 February 2024, pp. 1902-1914
- Print publication:
- 14 June 2024
-
- Article
-
- You have access
- HTML
- Export citation
Notes on Article Contributors
-
- Journal:
- Nineteenth-Century Music Review / Volume 20 / Issue 3 / December 2023
- Published online by Cambridge University Press:
- 22 February 2024, pp. 469-470
-
- Article
- Export citation
