Refine listing
Actions for selected content:
1419635 results in Open Access
JEH volume 84 issue 4 Cover and Front matter
-
- Journal:
- The Journal of Economic History / Volume 84 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 25 November 2024, pp. f1-f4
- Print publication:
- December 2024
-
- Article
-
- You have access
- Export citation
STATISTICAL ANALYSIS ON COMPLEXLY STRUCTURED DATA
-
- Journal:
- Bulletin of the Australian Mathematical Society / Volume 111 / Issue 1 / February 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 184-185
- Print publication:
- February 2025
-
- Article
-
- You have access
- HTML
- Export citation
Risk factors for healthcare-associated infection among patients hospitalized with COVID-19 infection
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 45 / Issue 12 / December 2024
- Published online by Cambridge University Press:
- 25 November 2024, pp. 1482-1483
- Print publication:
- December 2024
-
- Article
- Export citation
Sailing to Calanais: Monument Complexes and the Sea in the Neolithic of Western Scotland and Beyond
-
- Journal:
- Proceedings of the Prehistoric Society / Volume 90 / December 2024
- Published online by Cambridge University Press:
- 25 November 2024, pp. 253-277
- Print publication:
- December 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The planar 3-colorable subgroup ε of Thompson’s group F and its even part
- Part of
-
- Journal:
- Proceedings of the Edinburgh Mathematical Society / Volume 68 / Issue 1 / February 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 173-197
-
- Article
- Export citation
-
We study the planar 3-colorablesubgroup
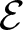 $\mathcal{E}$ of Thompson’s group F and its even part
$\mathcal{E}$ of Thompson’s group F and its even part 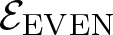 ${\mathcal{E}_{\rm EVEN}}$. The latter is obtained by cutting
${\mathcal{E}_{\rm EVEN}}$. The latter is obtained by cutting 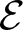 $\mathcal{E}$ with a finite index subgroup of F isomorphic to F, namely the rectangular subgroup
$\mathcal{E}$ with a finite index subgroup of F isomorphic to F, namely the rectangular subgroup 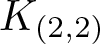 $K_{(2,2)}$. We show that the even part
$K_{(2,2)}$. We show that the even part 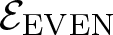 ${\mathcal{E}_{\rm EVEN}}$ of the planar 3-colorable subgroup admits a description in terms of stabilisers of suitable subsets of dyadic rationals. As a consequence
${\mathcal{E}_{\rm EVEN}}$ of the planar 3-colorable subgroup admits a description in terms of stabilisers of suitable subsets of dyadic rationals. As a consequence 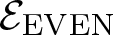 ${\mathcal{E}_{\rm EVEN}}$ is closed in the sense of Golan and Sapir. We then study three quasi-regular representations associated with
${\mathcal{E}_{\rm EVEN}}$ is closed in the sense of Golan and Sapir. We then study three quasi-regular representations associated with 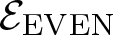 ${\mathcal{E}_{\rm EVEN}}$: two are shown to be irreducible and one to be reducible.
${\mathcal{E}_{\rm EVEN}}$: two are shown to be irreducible and one to be reducible.
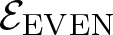
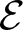
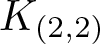
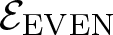
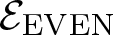
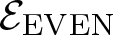
Destruction, delay and déjà vu: the restoration of Dublin’s O’Connell Street after the Irish civil war, 1922–1933
-
- Journal:
- Urban History / Volume 52 / Issue 4 / November 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 659-678
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Combining internet-delivered cognitive behavioural therapy and attention bias modification for reducing depressive symptoms in firefighters: a randomized controlled trial
-
- Journal:
- Behavioural and Cognitive Psychotherapy / Volume 53 / Issue 1 / March 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 63-73
- Print publication:
- March 2025
-
- Article
- Export citation
Aging in a Changing World: Older New Zealanders and Contemporary Multiculturalism, Molly George, Rutgers University Press, New Brunswick, NJ, 2022, 192 pp., pbk $32.95, ISBN 13: 9781978809406
-
- Journal:
- Ageing & Society / Volume 45 / Issue 5 / May 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 1034-1036
- Print publication:
- May 2025
-
- Article
- Export citation

Preserving large-scale features in simulations of elastic turbulence
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 25 November 2024, A37
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Christina Ramos, Bedlam in the New World: A Mexican Madhouse in the Age of Enlightenment (Chapel Hill: University of North Carolina Press, 2022), pp. xiv + 250, paperback, ISBN 978-146-9666-570.
-
- Journal:
- Medical History / Volume 68 / Issue 4 / October 2024
- Published online by Cambridge University Press:
- 25 November 2024, pp. 484-485
-
- Article
-
- You have access
- HTML
- Export citation

Existence of Bolgiano–Obukhov scaling in the bottom ocean?
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 25 November 2024, A38
-
- Article
- Export citation
-
The seminal Bolgiano–Obukhov (BO) theory established the fundamental framework for turbulent mixing and energy transfer in stably stratified fluids. However, the presence of BO scalings remains debatable despite their being observed in stably stratified atmospheric layers and convective turbulence. In this study, we performed precise temperature measurements with 51 high-resolution loggers above the seafloor for 46 h on the continental shelf of the northern South China Sea. The temperature observation exhibits three layers with increasing distance from the seafloor: the bottom mixed layer (BML), the mixing zone and the internal wave zone. A BO-like scaling
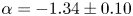 $\alpha =-1.34\pm 0.10$ is observed in the temperature spectrum when the BML is in a weakly stable stratified (
$\alpha =-1.34\pm 0.10$ is observed in the temperature spectrum when the BML is in a weakly stable stratified (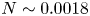 $N\sim 0.0018$ rad s
$N\sim 0.0018$ rad s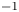 $^{-1}$) and strongly sheared (
$^{-1}$) and strongly sheared (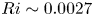 $Ri\sim 0.0027$) condition, whereas in the unstably stratified convective turbulence of the BML, the scaling
$Ri\sim 0.0027$) condition, whereas in the unstably stratified convective turbulence of the BML, the scaling 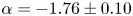 $\alpha =-1.76\pm 0.10$ clearly deviated from the BO theory but approached the classical
$\alpha =-1.76\pm 0.10$ clearly deviated from the BO theory but approached the classical  $-$5/3 scaling in isotropic turbulence. This suggests that the convective turbulence is not the promise of BO scaling. In the mixing zone, where internal waves alternately interact with the BML, the scaling follows the Kolmogorov scaling. In the internal wave zone, the scaling
$-$5/3 scaling in isotropic turbulence. This suggests that the convective turbulence is not the promise of BO scaling. In the mixing zone, where internal waves alternately interact with the BML, the scaling follows the Kolmogorov scaling. In the internal wave zone, the scaling 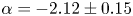 $\alpha =-2.12 \pm 0.15$ is observed in the turbulence range and possible mechanisms are provided.
$\alpha =-2.12 \pm 0.15$ is observed in the turbulence range and possible mechanisms are provided.
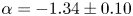
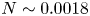
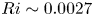
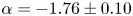

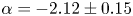

Turbulent/turbulent entrainment in a planar wake
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 25 November 2024, A26
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

A pore-scale resolved direct numerical simulation study for scaling analysis of the solutal convection in porous media
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 25 November 2024, A21
-
- Article
- Export citation
-
Understanding the solutal convection is a crucial step towards accurate prediction of CO
 $_2$ sequestration in deep saline aquifers. In this study, pore-scale resolved direct numerical simulations (DNS) are performed to analyse the scaling laws of the solutal convection in porous media. The porous media studied are composed of uniformly distributed square or circular elements. The Rayleigh numbers in the range
$_2$ sequestration in deep saline aquifers. In this study, pore-scale resolved direct numerical simulations (DNS) are performed to analyse the scaling laws of the solutal convection in porous media. The porous media studied are composed of uniformly distributed square or circular elements. The Rayleigh numbers in the range 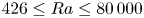 $426 \le Ra \le 80\,000$, the Darcy numbers in the range
$426 \le Ra \le 80\,000$, the Darcy numbers in the range 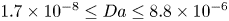 $1.7\times 10^{-8} \le Da \le 8.8\times 10^{-6}$ and the Schmidt numbers in the range
$1.7\times 10^{-8} \le Da \le 8.8\times 10^{-6}$ and the Schmidt numbers in the range 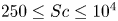 $250 \le Sc \le 10^4$ are considered in the DNS. The main time, length and velocity scales affecting the solutal convection are classified as the diffusive scales (
$250 \le Sc \le 10^4$ are considered in the DNS. The main time, length and velocity scales affecting the solutal convection are classified as the diffusive scales (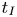 $t_I$,
$t_I$, 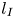 $l_I$ and
$l_I$ and 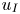 $u_I$), the convective scales (
$u_I$), the convective scales (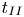 $t_{II}$,
$t_{II}$, 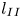 $l_{II}$ and
$l_{II}$ and 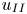 $u_{II}$) and the shut-down scales (
$u_{II}$) and the shut-down scales (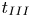 $t_{III}$,
$t_{III}$, 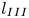 $l_{III}$ and
$l_{III}$ and 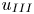 $u_{III}$). These scales determine the pore-scale Rayleigh number
$u_{III}$). These scales determine the pore-scale Rayleigh number 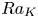 $Ra_K$ and the Rayleigh number
$Ra_K$ and the Rayleigh number 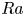 $Ra$. Based on the DNS results, the scaling laws for the transient dissolution flux are proposed in the different regimes of convection. It is found that the onset time of convection (
$Ra$. Based on the DNS results, the scaling laws for the transient dissolution flux are proposed in the different regimes of convection. It is found that the onset time of convection (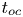 $t_{oc}$) and the period of the flux-growth regime (
$t_{oc}$) and the period of the flux-growth regime (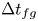 $\Delta t_{fg}$) vary linearly with the convective time scale
$\Delta t_{fg}$) vary linearly with the convective time scale 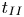 $t_{II}$. The merging of the original plumes and the re-initiation of the new plumes start in the same period, resulting in the merging re-initiation regime. Horizontal dispersion plays an important role in plume merging. The dissolution flux
$t_{II}$. The merging of the original plumes and the re-initiation of the new plumes start in the same period, resulting in the merging re-initiation regime. Horizontal dispersion plays an important role in plume merging. The dissolution flux 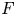 $F$ and the time since the onset of convection
$F$ and the time since the onset of convection 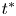 $t^{\ast }$ have an
$t^{\ast }$ have an 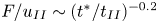 $F / u_{II} \sim (t^{\ast }/ t_{II})^{-0.2}$ scaling in the later stage of the merging re-initiation regime. This scaling is caused by the merging of the low-wavenumber plumes. It becomes more pronounced with decreasing porosity and leads to the nonlinear relationship between the Sherwood number
$F / u_{II} \sim (t^{\ast }/ t_{II})^{-0.2}$ scaling in the later stage of the merging re-initiation regime. This scaling is caused by the merging of the low-wavenumber plumes. It becomes more pronounced with decreasing porosity and leads to the nonlinear relationship between the Sherwood number 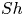 $Sh$ and
$Sh$ and 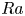 $Ra$ when the domain is not high enough for the plumes to fully develop. According to the DNS results, a regime is expected that is independent of both
$Ra$ when the domain is not high enough for the plumes to fully develop. According to the DNS results, a regime is expected that is independent of both 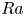 $Ra$ and
$Ra$ and 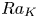 $Ra_K$, while the dimensionless constant flux
$Ra_K$, while the dimensionless constant flux 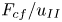 $F_{cf}/u_{II}$ in this regime decreases with decreasing porosity. When the mean solute concentration reaches approximately 30 %, convection enters the shut-down regime. For large
$F_{cf}/u_{II}$ in this regime decreases with decreasing porosity. When the mean solute concentration reaches approximately 30 %, convection enters the shut-down regime. For large 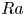 $Ra$, the dimensionless flux in the shut-down regime follows the scaling law
$Ra$, the dimensionless flux in the shut-down regime follows the scaling law 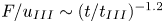 $F/u_{III}\sim (t/t_{III})^{-1.2}$ at large porosity (
$F/u_{III}\sim (t/t_{III})^{-1.2}$ at large porosity (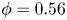 $\phi =0.56$) and
$\phi =0.56$) and 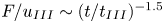 $F/u_{III}\sim (t/t_{III})^{-1.5}$ at small porosity (
$F/u_{III}\sim (t/t_{III})^{-1.5}$ at small porosity (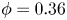 $\phi =0.36$ or
$\phi =0.36$ or 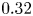 $0.32$). The study shows the significant pore-scale effect on the convection. The DNS cases in this study are mainly for simplified geometries and large
$0.32$). The study shows the significant pore-scale effect on the convection. The DNS cases in this study are mainly for simplified geometries and large 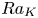 $Ra_K$. This can lead to uncertainties of the obtained scaling coefficients. It is important to determine the scaling coefficients for real geological formations to predict a real CO
$Ra_K$. This can lead to uncertainties of the obtained scaling coefficients. It is important to determine the scaling coefficients for real geological formations to predict a real CO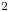 $_2$ sequestration process.
$_2$ sequestration process.
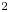
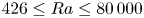
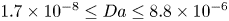
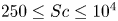
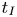
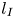
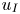
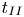
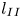
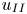
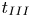
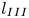
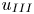
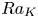
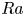
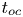
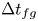
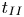
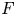
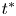
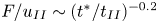
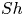
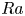
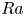
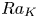
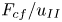
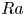
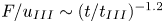
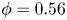
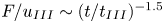
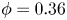
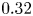
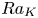
BOUNDEDNESS OF DIFFERENTIAL TRANSFORMS FOR FRACTIONAL HEAT SEMIGROUPS GENERATED BY SCHRÖDINGER OPERATORS
- Part of
-
- Journal:
- Journal of the Australian Mathematical Society / Volume 118 / Issue 2 / April 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 210-244
- Print publication:
- April 2025
-
- Article
- Export citation
-
Let
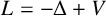 $L=-\Delta +V$ be a Schrödinger operator in
$L=-\Delta +V$ be a Schrödinger operator in 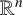 ${\mathbb R}^n$ with
${\mathbb R}^n$ with 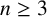 $n\geq 3$, where
$n\geq 3$, where 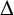 $\Delta $ is the Laplace operator denoted by
$\Delta $ is the Laplace operator denoted by 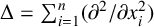 $\Delta =\sum ^{n}_{i=1}({\partial ^{2}}/{\partial x_{i}^{2}})$ and the nonnegative potential V belongs to the reverse Hölder class
$\Delta =\sum ^{n}_{i=1}({\partial ^{2}}/{\partial x_{i}^{2}})$ and the nonnegative potential V belongs to the reverse Hölder class  $(RH)_{q}$ with
$(RH)_{q}$ with 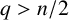 $q>n/2$. For
$q>n/2$. For 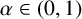 $\alpha \in (0,1)$, we define the operator
$\alpha \in (0,1)$, we define the operator 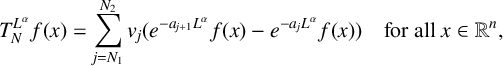 $$ \begin{align*} T_N^{L^{\alpha}} f(x) =\sum_{j=N_1}^{N_2} v_j(e^{-a_{j+1}L^\alpha} f(x)-e^{-a_{j}L^\alpha} f(x)) \quad \mbox{for all }x\in \mathbb R^n, \end{align*} $$
$$ \begin{align*} T_N^{L^{\alpha}} f(x) =\sum_{j=N_1}^{N_2} v_j(e^{-a_{j+1}L^\alpha} f(x)-e^{-a_{j}L^\alpha} f(x)) \quad \mbox{for all }x\in \mathbb R^n, \end{align*} $$where
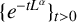 $\{e^{-tL^\alpha } \}_{t>0}$ is the fractional heat semigroup of the operator L,
$\{e^{-tL^\alpha } \}_{t>0}$ is the fractional heat semigroup of the operator L, 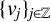 $\{v_j\}_{j\in \mathbb Z}$ is a bounded real sequence and
$\{v_j\}_{j\in \mathbb Z}$ is a bounded real sequence and 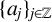 $\{a_j\}_{j\in \mathbb Z}$ is an increasing real sequence.
$\{a_j\}_{j\in \mathbb Z}$ is an increasing real sequence.We investigate the boundedness of the operator
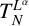 $T_N^{L^{\alpha }}$ and the related maximal operator
$T_N^{L^{\alpha }}$ and the related maximal operator 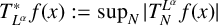 $T^*_{L^{\alpha }}f(x):=\sup _N \vert T_N^{L^{\alpha }} f(x)\vert $ on the spaces
$T^*_{L^{\alpha }}f(x):=\sup _N \vert T_N^{L^{\alpha }} f(x)\vert $ on the spaces 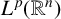 $L^{p}(\mathbb {R}^{n})$ and
$L^{p}(\mathbb {R}^{n})$ and 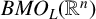 $BMO_{L}(\mathbb {R}^{n})$, respectively. As extensions of
$BMO_{L}(\mathbb {R}^{n})$, respectively. As extensions of 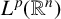 $L^{p}(\mathbb {R}^{n})$, the boundedness of the operators
$L^{p}(\mathbb {R}^{n})$, the boundedness of the operators 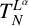 $T_N^{L^{\alpha }}$ and
$T_N^{L^{\alpha }}$ and 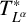 $T^*_{L^{\alpha }}$ on the Morrey space
$T^*_{L^{\alpha }}$ on the Morrey space 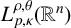 $L^{\rho ,\theta }_{p,\kappa }(\mathbb {R}^{n})$ and the weak Morrey space
$L^{\rho ,\theta }_{p,\kappa }(\mathbb {R}^{n})$ and the weak Morrey space 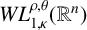 $WL^{\rho ,\theta }_{1,\kappa }(\mathbb {R}^{n})$ has also been proved.
$WL^{\rho ,\theta }_{1,\kappa }(\mathbb {R}^{n})$ has also been proved.
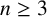
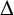
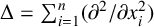

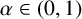
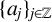
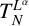
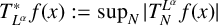
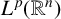
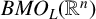
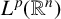
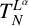
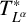
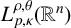
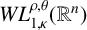
Random growth via gradient flow aggregation
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 62 / Issue 2 / June 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 735-755
- Print publication:
- June 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
We introduce gradient flow aggregation, a random growth model. Given existing particles
 $\{x_1,\ldots,x_n\} \subset \mathbb{R}^2$, a new particle arrives from a random direction at
$\{x_1,\ldots,x_n\} \subset \mathbb{R}^2$, a new particle arrives from a random direction at  $\infty$ and flows in direction of the vector field
$\infty$ and flows in direction of the vector field  $\nabla E$ where
$\nabla E$ where  $ E(x) = \sum_{i=1}^{n}{1}/{\|x-x_i\|^{\alpha}}$,
$ E(x) = \sum_{i=1}^{n}{1}/{\|x-x_i\|^{\alpha}}$,  $0 < \alpha < \infty$. The case
$0 < \alpha < \infty$. The case  $\alpha = 0$ refers to the logarithmic energy
$\alpha = 0$ refers to the logarithmic energy  ${-}\sum\log\|x-x_i\|$. Particles stop once they are at distance 1 from one of the existing particles, at which point they are added to the set and remain fixed for all time. We prove, under a non-degeneracy assumption, a Beurling-type estimate which, via Kesten’s method, can be used to deduce sub-ballistic growth for
${-}\sum\log\|x-x_i\|$. Particles stop once they are at distance 1 from one of the existing particles, at which point they are added to the set and remain fixed for all time. We prove, under a non-degeneracy assumption, a Beurling-type estimate which, via Kesten’s method, can be used to deduce sub-ballistic growth for  $0 \leq \alpha < 1$,
$0 \leq \alpha < 1$,  $\text{diam}(\{x_1,\ldots,x_n\}) \leq c_{\alpha} \cdot n^{({3 \alpha +1})/({2\alpha + 2})}$. This is optimal when
$\text{diam}(\{x_1,\ldots,x_n\}) \leq c_{\alpha} \cdot n^{({3 \alpha +1})/({2\alpha + 2})}$. This is optimal when  $\alpha = 0$. The case
$\alpha = 0$. The case  $\alpha = 0$ leads to a ‘round’ full-dimensional tree. The larger the value of
$\alpha = 0$ leads to a ‘round’ full-dimensional tree. The larger the value of  $\alpha$, the sparser the tree. Some instances of the higher-dimensional setting are also discussed.
$\alpha$, the sparser the tree. Some instances of the higher-dimensional setting are also discussed.













Instability evolution on a shock-accelerated cylindrical fluid layer with arbitrary Atwood numbers
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 25 November 2024, A9
-
- Article
- Export citation
-
Developing a model to describe the shock-accelerated cylindrical fluid layer with arbitrary Atwood numbers is essential for uncovering the effect of Atwood numbers on the perturbation growth. The recent model (J. Fluid Mech., vol. 969, 2023, p. A6) reveals several contributions to the instability evolution of a shock-accelerated cylindrical fluid layer but its applicability is limited to cases with an absolute value of Atwood numbers close to
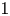 $1$, due to the employment of the thin-shell correction and interface coupling effect of the fluid layer in vacuum. By employing the linear stability analysis on a cylindrical fluid layer in which two interfaces separate three arbitrary-density fluids, the present work generalizes the thin-shell correction and interface coupling effect, and thus, extends the recent model to cases with arbitrary Atwood numbers. The accuracy of this extended model in describing the instability evolution of the shock-accelerated fluid layer before reshock is confirmed via direct numerical simulations. In the verification simulations, three fluid-layer configurations are considered, where the outer and intermediate fluids remain fixed and the density of the inner fluid is reduced. Moreover, the mechanisms underlying the effect of the Atwood number at the inner interface on the perturbation growth are mainly elucidated by employing the model to analyse each contribution. As the Atwood number decreases, the dominant contribution of the Richtmyer–Meshkov instability is enhanced due to the stronger waves reverberated inside the layer, leading to weakened perturbation growth at initial in-phase interfaces and enhanced perturbation growth at initial anti-phase interfaces.
$1$, due to the employment of the thin-shell correction and interface coupling effect of the fluid layer in vacuum. By employing the linear stability analysis on a cylindrical fluid layer in which two interfaces separate three arbitrary-density fluids, the present work generalizes the thin-shell correction and interface coupling effect, and thus, extends the recent model to cases with arbitrary Atwood numbers. The accuracy of this extended model in describing the instability evolution of the shock-accelerated fluid layer before reshock is confirmed via direct numerical simulations. In the verification simulations, three fluid-layer configurations are considered, where the outer and intermediate fluids remain fixed and the density of the inner fluid is reduced. Moreover, the mechanisms underlying the effect of the Atwood number at the inner interface on the perturbation growth are mainly elucidated by employing the model to analyse each contribution. As the Atwood number decreases, the dominant contribution of the Richtmyer–Meshkov instability is enhanced due to the stronger waves reverberated inside the layer, leading to weakened perturbation growth at initial in-phase interfaces and enhanced perturbation growth at initial anti-phase interfaces.
Uniqueness theorems for meromorphic inner functions and canonical systems
- Part of
-
- Journal:
- Canadian Mathematical Bulletin / Volume 68 / Issue 1 / March 2025
- Published online by Cambridge University Press:
- 25 November 2024, pp. 124-140
- Print publication:
- March 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Identification of cross-frequency interactions in compressible cavity flow using harmonic resolvent analysis
-
- Journal:
- Journal of Fluid Mechanics / Volume 1000 / 10 December 2024
- Published online by Cambridge University Press:
- 25 November 2024, A13
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The resolvent analysis reveals the worst-case disturbances and the most amplified response in a fluid flow that can develop around a stationary base state. The recent work by Padovan et al. (J. Fluid Mech., vol. 900, 2020, A14) extended the classical resolvent analysis to the harmonic resolvent analysis framework by incorporating the time-varying nature of the base flow. The harmonic resolvent analysis can capture the triadic interactions between perturbations at two different frequencies through a base flow at a particular frequency. The singular values of the harmonic resolvent operator act as a gain between the spatiotemporal forcing and the response provided by the singular vectors. In the current study, we formulate the harmonic resolvent analysis framework for compressible flows based on the linearized Navier–Stokes equation (i.e. operator-based formulation). We validate our approach by applying the technique to the low-Mach-number flow past an airfoil. We further illustrate the application of this method to compressible cavity flows at Mach numbers of 0.6 and 0.8 with a length-to-depth ratio of
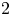 $2$. For the cavity flow at a Mach number of 0.6, the harmonic resolvent analysis reveals that the nonlinear cross-frequency interactions dominate the amplification of perturbations at frequencies that are harmonics of the leading Rossiter mode in the nonlinear flow. The findings demonstrate a physically consistent representation of an energy transfer from slow-evolving modes toward fast-evolving modes in the flow through cross-frequency interactions. For the cavity flow at a Mach number of 0.8, the analysis also sheds light on the nature of cross-frequency interaction in a cavity flow with two coexisting resonances.
$2$. For the cavity flow at a Mach number of 0.6, the harmonic resolvent analysis reveals that the nonlinear cross-frequency interactions dominate the amplification of perturbations at frequencies that are harmonics of the leading Rossiter mode in the nonlinear flow. The findings demonstrate a physically consistent representation of an energy transfer from slow-evolving modes toward fast-evolving modes in the flow through cross-frequency interactions. For the cavity flow at a Mach number of 0.8, the analysis also sheds light on the nature of cross-frequency interaction in a cavity flow with two coexisting resonances.
Navigating Diphtheria Resurgence in Pakistan’s Conflict-Ridden and Disaster-Prone Area
-
- Journal:
- Disaster Medicine and Public Health Preparedness / Volume 18 / 2024
- Published online by Cambridge University Press:
- 25 November 2024, e288
-
- Article
- Export citation
