Refine listing
Actions for selected content:
1418449 results in Open Access
A HIERARCHY ON NON-ARCHIMEDEAN POLISH GROUPS ADMITTING A COMPATIBLE COMPLETE LEFT-INVARIANT METRIC
- Part of
-
- Journal:
- The Journal of Symbolic Logic , First View
- Published online by Cambridge University Press:
- 06 February 2024, pp. 1-19
-
- Article
- Export citation
-
In this article, we introduce a hierarchy on the class of non-archimedean Polish groups that admit a compatible complete left-invariant metric. We denote this hierarchy by
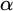 $\alpha $-CLI and L-
$\alpha $-CLI and L-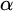 $\alpha $-CLI where
$\alpha $-CLI where 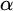 $\alpha $ is a countable ordinal. We establish three results:
$\alpha $ is a countable ordinal. We establish three results: (1) G is
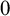 $0$-CLI iff
$0$-CLI iff 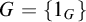 $G=\{1_G\}$;
$G=\{1_G\}$;(2) G is
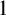 $1$-CLI iff G admits a compatible complete two-sided invariant metric; and
$1$-CLI iff G admits a compatible complete two-sided invariant metric; and(3) G is L-
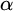 $\alpha $-CLI iff G is locally
$\alpha $-CLI iff G is locally 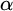 $\alpha $-CLI, i.e., G contains an open subgroup that is
$\alpha $-CLI, i.e., G contains an open subgroup that is 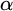 $\alpha $-CLI.
$\alpha $-CLI.
Subsequently, we show this hierarchy is proper by constructing non-archimedean CLI Polish groups
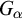 $G_\alpha $ and
$G_\alpha $ and 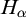 $H_\alpha $ for
$H_\alpha $ for 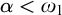 $\alpha <\omega _1$, such that:
$\alpha <\omega _1$, such that: (1)
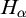 $H_\alpha $ is
$H_\alpha $ is 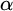 $\alpha $-CLI but not L-
$\alpha $-CLI but not L-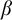 $\beta $-CLI for
$\beta $-CLI for 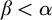 $\beta <\alpha $; and
$\beta <\alpha $; and(2)
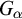 $G_\alpha $ is
$G_\alpha $ is 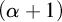 $(\alpha +1)$-CLI but not L-
$(\alpha +1)$-CLI but not L-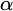 $\alpha $-CLI.
$\alpha $-CLI.
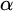
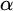
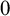
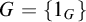
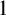
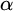
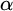
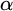
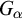
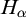
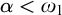
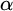
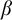
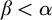
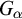
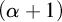
Lettere e Documenti / Pisma in Dokumenti / Letters and Documents, two volumes Giuseppe Tartini (1692–1770), ed. Giorgia Malagò Trieste: Edizioni Università di Trieste, 2020 Volume 1, pp. 374, ISBN 978 8 855 11066 2 Volume 2, pp. 531, ISBN 978 8 855 11068 6
-
- Journal:
- Eighteenth-Century Music / Volume 21 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 06 February 2024, pp. 89-91
- Print publication:
- March 2024
-
- Article
- Export citation
ESO volume 25 issue 1 Cover and Front matter
-
- Journal:
- Enterprise & Society / Volume 25 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 06 February 2024, pp. f1-f5
- Print publication:
- March 2024
-
- Article
-
- You have access
- Export citation

Covering convection with a thermal blanket: numerical simulation and stochastic modelling
-
- Journal:
- Journal of Fluid Mechanics / Volume 980 / 10 February 2024
- Published online by Cambridge University Press:
- 06 February 2024, A47
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Philon und Theone Georg Anton Benda (1722–1795), ed. Austin Glatthorn Middleton, WI: A-R Editions, 2020 pp. xxiv + 166, ISBN 978 1 9872 0456 8
-
- Journal:
- Eighteenth-Century Music / Volume 21 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 06 February 2024, pp. 79-83
- Print publication:
- March 2024
-
- Article
- Export citation
The causes of wheat stem sawfly (Hymenoptera: Cephidae) larval mortality in the Canadian prairies
-
- Journal:
- The Canadian Entomologist / Volume 156 / 2024
- Published online by Cambridge University Press:
- 06 February 2024, e3
-
- Article
- Export citation
RADIOCARBON AND URANIUM PROFILES IN MARINE GASTROPODS AROUND THE JAPANESE ARCHIPELAGO
-
- Journal:
- Radiocarbon / Volume 66 / Issue 6 / December 2024
- Published online by Cambridge University Press:
- 06 February 2024, pp. 1883-1897
- Print publication:
- December 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Supergranule aggregation: a Prandtl number-independent feature of constant heat flux-driven convection flows
-
- Journal:
- Journal of Fluid Mechanics / Volume 980 / 10 February 2024
- Published online by Cambridge University Press:
- 06 February 2024, A46
-
- Article
- Export citation
-
Supergranule aggregation, i.e. the gradual aggregation of convection cells to horizontally extended networks of flow structures, is a unique feature of constant heat flux-driven turbulent convection. In the present study, we address the question if this mechanism of self-organisation of the flow is present for any fluid. Therefore, we analyse three-dimensional Rayleigh–Bénard convection at a fixed Rayleigh number
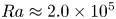 ${Ra} \approx 2.0 \times 10^{5}$ across
${Ra} \approx 2.0 \times 10^{5}$ across 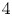 $4$ orders of Prandtl numbers
$4$ orders of Prandtl numbers 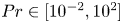 ${Pr} \in [10^{-2}, 10^{2}]$ by means of direct numerical simulations in horizontally extended periodic domains with aspect ratio
${Pr} \in [10^{-2}, 10^{2}]$ by means of direct numerical simulations in horizontally extended periodic domains with aspect ratio  $\varGamma = 60$. Our study confirms the omnipresence of the mechanism of supergranule aggregation for the entire range of investigated fluids. Moreover, we analyse the effect of
$\varGamma = 60$. Our study confirms the omnipresence of the mechanism of supergranule aggregation for the entire range of investigated fluids. Moreover, we analyse the effect of 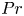 ${Pr}$ on the global heat and momentum transport, and clarify the role of a potential stable stratification in the bulk of the fluid layer. The ubiquity of the investigated mechanism of flow self-organisation underlines its relevance for pattern formation in geophysical and astrophysical convection flows, the latter of which are often driven by prescribed heat fluxes.
${Pr}$ on the global heat and momentum transport, and clarify the role of a potential stable stratification in the bulk of the fluid layer. The ubiquity of the investigated mechanism of flow self-organisation underlines its relevance for pattern formation in geophysical and astrophysical convection flows, the latter of which are often driven by prescribed heat fluxes.
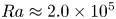
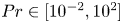

Feminist Politics in Neoconservative Russia: An Ethnography of Resistance and Resources. By Inna Perheentupa. Bristol: Bristol University Press. 204 pp. GBP 80. ISBN 978-1529216967.
-
- Journal:
- Politics & Gender / Volume 20 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 06 February 2024, pp. 1022-1024
-
- Article
- Export citation
SMOKE ON THE WATER: HES AT 50 AND THE NON-NEUTRALITY OF HISTORY
-
- Journal:
- Journal of the History of Economic Thought / Volume 46 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 06 February 2024, pp. 568-575
- Print publication:
- December 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The Mini Nutritional Assessment combined with body fat for detecting the risk of sarcopenia and sarcopenic obesity in metabolic syndrome
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 10 / 28 May 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. 1659-1667
- Print publication:
- 28 May 2024
-
- Article
-
- You have access
- HTML
- Export citation
Why Latin American Parties Are Not Coming Back
-
- Journal:
- Latin American Politics and Society / Volume 66 / Issue 3 / August 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. 164-193
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
ETS volume 44 issue 3 Cover and Front matter
-
- Journal:
- Ergodic Theory and Dynamical Systems / Volume 44 / Issue 3 / March 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. f1-f2
- Print publication:
- March 2024
-
- Article
-
- You have access
- Export citation
The Earth Is Sweet. On Cottica Ndyuka (De)compositions
-
- Journal:
- Comparative Studies in Society and History / Volume 66 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. 242-266
-
- Article
- Export citation
A Matter of Opinion? How Unexpected Opinion Authors Influence Support for Supreme Court Decisions
-
- Journal:
- Journal of Law and Courts / Volume 12 / Issue 1 / April 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. 144-164
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Impromptu Sarah O'Brien, harp Audite, 97.807, 2022 (1 CD: 80 minutes) €19.99
-
- Journal:
- Nineteenth-Century Music Review / Volume 21 / Issue 3 / December 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. 583-588
-
- Article
- Export citation
Gyan Prakash and Jeremy Adelman, eds., Inventing the Third World: In Search of Freedom for the Postwar Global South, Histories of Internationalism (London: Bloomsbury Academic, 2023), pp. 256, $159.95 (hardcover). ISBN: 9781350268159.
-
- Journal:
- Journal of the History of Economic Thought / Volume 46 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. 481-484
- Print publication:
- September 2024
-
- Article
- Export citation
Canadian Consensus Guidelines for the Diagnosis and Treatment of Autoimmune Encephalitis in Adults
-
- Journal:
- Canadian Journal of Neurological Sciences / Volume 51 / Issue 6 / November 2024
- Published online by Cambridge University Press:
- 05 February 2024, pp. 734-754
-
- Article
-
- You have access
- Open access
- HTML
- Export citation

Self-similarity of the dipole–multipole transition in rapidly rotating dynamos
-
- Journal:
- Journal of Fluid Mechanics / Volume 980 / 10 February 2024
- Published online by Cambridge University Press:
- 05 February 2024, A30
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
