Refine search
Actions for selected content:
53194 results in Statistics and Probability
Global and local evolutionary dynamics of Dengue virus serotypes 1, 3, and 4
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 09 June 2023, e127
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Data collaborations at a local scale: Lessons learnt in Rennes (2010–2021)
-
- Journal:
- Data & Policy / Volume 5 / 2023
- Published online by Cambridge University Press:
- 09 June 2023, e20
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
On the number of error correcting codes
- Part of
-
- Journal:
- Combinatorics, Probability and Computing / Volume 32 / Issue 5 / September 2023
- Published online by Cambridge University Press:
- 09 June 2023, pp. 819-832
-
- Article
- Export citation
-
We show that for a fixed
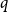 $q$, the number of
$q$, the number of 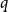 $q$-ary
$q$-ary 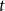 $t$-error correcting codes of length
$t$-error correcting codes of length  $n$ is at most
$n$ is at most 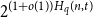 $2^{(1 + o(1)) H_q(n,t)}$ for all
$2^{(1 + o(1)) H_q(n,t)}$ for all 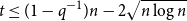 $t \leq (1 - q^{-1})n - 2\sqrt{n \log n}$, where
$t \leq (1 - q^{-1})n - 2\sqrt{n \log n}$, where 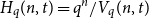 $H_q(n, t) = q^n/ V_q(n,t)$ is the Hamming bound and
$H_q(n, t) = q^n/ V_q(n,t)$ is the Hamming bound and 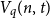 $V_q(n,t)$ is the cardinality of the radius
$V_q(n,t)$ is the cardinality of the radius 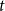 $t$ Hamming ball. This proves a conjecture of Balogh, Treglown, and Wagner, who showed the result for
$t$ Hamming ball. This proves a conjecture of Balogh, Treglown, and Wagner, who showed the result for 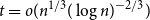 $t = o(n^{1/3} (\log n)^{-2/3})$.
$t = o(n^{1/3} (\log n)^{-2/3})$.

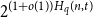
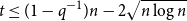
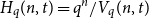
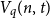
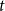
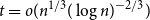
Coinfection and repeat bacterial sexually transmitted infections (STI) – retrospective study on male attendees of public STI clinics in an Asia Pacific city
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 09 June 2023, e101
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Different epidemiological characteristics between patients with non-hospital-onset and hospital-onset candidemia: a retrospective cohort study
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 09 June 2023, e102
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Spanning
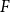 $F$-cycles in random graphs
$F$-cycles in random graphs
- Part of
-
- Journal:
- Combinatorics, Probability and Computing / Volume 32 / Issue 5 / September 2023
- Published online by Cambridge University Press:
- 09 June 2023, pp. 833-850
-
- Article
- Export citation
-
We extend a recent argument of Kahn, Narayanan and Park ((2021) Proceedings of the AMS 149 3201–3208) about the threshold for the appearance of the square of a Hamilton cycle to other spanning structures. In particular, for any spanning graph, we give a sufficient condition under which we may determine its threshold. As an application, we find the threshold for a set of cyclically ordered copies of
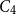 $C_4$ that span the entire vertex set, so that any two consecutive copies overlap in exactly one edge and all overlapping edges are disjoint. This answers a question of Frieze. We also determine the threshold for edge-overlapping spanning
$C_4$ that span the entire vertex set, so that any two consecutive copies overlap in exactly one edge and all overlapping edges are disjoint. This answers a question of Frieze. We also determine the threshold for edge-overlapping spanning 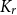 $K_r$-cycles.
$K_r$-cycles.
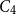
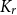
Evaluating probabilistic forecasts for maritime engineering operations
- Part of
-
- Journal:
- Data-Centric Engineering / Volume 4 / 2023
- Published online by Cambridge University Press:
- 09 June 2023, e15
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Unimodular random one-ended planar graphs are sofic
- Part of
-
- Journal:
- Combinatorics, Probability and Computing / Volume 32 / Issue 6 / November 2023
- Published online by Cambridge University Press:
- 09 June 2023, pp. 851-858
-
- Article
- Export citation
A smoother notion of spread hypergraphs
- Part of
-
- Journal:
- Combinatorics, Probability and Computing / Volume 32 / Issue 5 / September 2023
- Published online by Cambridge University Press:
- 08 June 2023, pp. 809-818
-
- Article
- Export citation
-
Alweiss, Lovett, Wu, and Zhang introduced
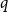 $q$-spread hypergraphs in their breakthrough work regarding the sunflower conjecture, and since then
$q$-spread hypergraphs in their breakthrough work regarding the sunflower conjecture, and since then 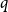 $q$-spread hypergraphs have been used to give short proofs of several outstanding problems in probabilistic combinatorics. A variant of
$q$-spread hypergraphs have been used to give short proofs of several outstanding problems in probabilistic combinatorics. A variant of 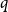 $q$-spread hypergraphs was implicitly used by Kahn, Narayanan, and Park to determine the threshold for when a square of a Hamiltonian cycle appears in the random graph
$q$-spread hypergraphs was implicitly used by Kahn, Narayanan, and Park to determine the threshold for when a square of a Hamiltonian cycle appears in the random graph 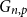 $G_{n,p}$. In this paper, we give a common generalization of the original notion of
$G_{n,p}$. In this paper, we give a common generalization of the original notion of 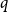 $q$-spread hypergraphs and the variant used by Kahn, Narayanan, and Park.
$q$-spread hypergraphs and the variant used by Kahn, Narayanan, and Park.
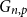
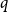
Reach and uptake of mass drug administration for worm infections through health facility-, school-, and community-based approaches in two districts of Zambia: a call for scale-up
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 08 June 2023, e183
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Fermenting a place in history: The first outbreak of Escherichia coli O157 associated with kimchi in Canada
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 08 June 2023, e106
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Association of patient characteristics with clinical outcomes in a cohort of hospitalised patients with SARS-CoV-2 infection in a Greek referral centre for COVID-19 – Corrigendum
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 08 June 2023, e91
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Cyber insurance-linked securities
-
- Journal:
- ASTIN Bulletin: The Journal of the IAA / Volume 53 / Issue 3 / September 2023
- Published online by Cambridge University Press:
- 08 June 2023, pp. 684-705
- Print publication:
- September 2023
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
On a wider class of prior distributions for graphical models
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 08 June 2023, pp. 230-243
- Print publication:
- March 2024
-
- Article
- Export citation
-
Gaussian graphical models are useful tools for conditional independence structure inference of multivariate random variables. Unfortunately, Bayesian inference of latent graph structures is challenging due to exponential growth of
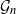 $\mathcal{G}_n$, the set of all graphs in n vertices. One approach that has been proposed to tackle this problem is to limit search to subsets of
$\mathcal{G}_n$, the set of all graphs in n vertices. One approach that has been proposed to tackle this problem is to limit search to subsets of 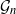 $\mathcal{G}_n$. In this paper we study subsets that are vector subspaces with the cycle space
$\mathcal{G}_n$. In this paper we study subsets that are vector subspaces with the cycle space 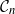 $\mathcal{C}_n$ as the main example. We propose a novel prior on
$\mathcal{C}_n$ as the main example. We propose a novel prior on 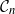 $\mathcal{C}_n$ based on linear combinations of cycle basis elements and present its theoretical properties. Using this prior, we implement a Markov chain Monte Carlo algorithm, and show that (i) posterior edge inclusion estimates computed with our technique are comparable to estimates from the standard technique despite searching a smaller graph space, and (ii) the vector space perspective enables straightforward implementation of MCMC algorithms.
$\mathcal{C}_n$ based on linear combinations of cycle basis elements and present its theoretical properties. Using this prior, we implement a Markov chain Monte Carlo algorithm, and show that (i) posterior edge inclusion estimates computed with our technique are comparable to estimates from the standard technique despite searching a smaller graph space, and (ii) the vector space perspective enables straightforward implementation of MCMC algorithms.
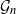
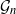
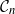
Bottom-up forecasting: Applications and limitations in load forecasting using smart-meter data
-
- Journal:
- Data-Centric Engineering / Volume 4 / 2023
- Published online by Cambridge University Press:
- 07 June 2023, e14
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
A scoping review of factors associated with antimicrobial-resistant Campylobacter species infections in humans
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 07 June 2023, e100
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Measuring the suboptimality of dividend controls in a Brownian risk model
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 55 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 07 June 2023, pp. 1442-1472
- Print publication:
- December 2023
-
- Article
- Export citation
Opening industry data: The private sector’s role in addressing societal challenges
-
- Journal:
- Data & Policy / Volume 5 / 2023
- Published online by Cambridge University Press:
- 07 June 2023, e19
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Exact recovery of Granger causality graphs with unconditional pairwise tests
-
- Journal:
- Network Science / Volume 11 / Issue 3 / September 2023
- Published online by Cambridge University Press:
- 06 June 2023, pp. 431-457
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Extrema of a multinomial assignment process
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 06 June 2023, pp. 198-213
- Print publication:
- March 2024
-
- Article
- Export citation
