Refine search
Actions for selected content:
53194 results in Statistics and Probability
Hepatitis B antibody levels after different doses of hepatitis B vaccination: a retrospective study based on hospitalized children
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 26 October 2023, e186
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Attribution of nosocomial seeding to long-term care facility COVID-19 outbreaks
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 25 October 2023, e191
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Using SNP addresses for Salmonella Typhimurium DT104 in routine veterinary outbreak detection
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 25 October 2023, e187
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Boolean percolation on digraphs and random exchange processes
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 3 / September 2024
- Published online by Cambridge University Press:
- 25 October 2023, pp. 755-766
- Print publication:
- September 2024
-
- Article
- Export citation
-
We study in a general graph-theoretic formulation a long-range percolation model introduced by Lamperti [27]. For various underlying digraphs, we discuss connections between this model and random exchange processes. We clarify, for all
 $n \in \mathbb{N}$, under which conditions the lattices
$n \in \mathbb{N}$, under which conditions the lattices  $\mathbb{N}_0^n$ and
$\mathbb{N}_0^n$ and  $\mathbb{Z}^n$ are essentially covered in this model. Moreover, for all
$\mathbb{Z}^n$ are essentially covered in this model. Moreover, for all 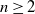 $n \geq 2$, we establish that it is impossible to cover the directed n-ary tree in our model.
$n \geq 2$, we establish that it is impossible to cover the directed n-ary tree in our model.



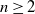
The role of sustainability knowledge-action platforms in advancing multi-stakeholder engagement on sustainability – ERRATUM
-
- Journal:
- Data & Policy / Volume 5 / 2023
- Published online by Cambridge University Press:
- 25 October 2023, e34
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
An inaccuracy measure between non-explosive point processes with applications to Markov chains
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 56 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 25 October 2023, pp. 735-756
- Print publication:
- June 2024
-
- Article
- Export citation
A changing climate for actuarial science
-
- Journal:
- Annals of Actuarial Science / Volume 17 / Issue 3 / November 2023
- Published online by Cambridge University Press:
- 24 October 2023, pp. 415-419
-
- Article
-
- You have access
- HTML
- Export citation
Bayesian system identification for structures considering spatial and temporal correlation
-
- Journal:
- Data-Centric Engineering / Volume 4 / 2023
- Published online by Cambridge University Press:
- 23 October 2023, e22
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Neural network ensembles and uncertainty estimation for predictions of inelastic mechanical deformation using a finite element method-neural network approach
- Part of
-
- Journal:
- Data-Centric Engineering / Volume 4 / 2023
- Published online by Cambridge University Press:
- 23 October 2023, e23
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
NONPARAMETRIC TIME-VARYING PANEL DATA MODELS WITH HETEROGENEITY
-
- Journal:
- Econometric Theory / Volume 41 / Issue 2 / April 2025
- Published online by Cambridge University Press:
- 23 October 2023, pp. 302-325
-
- Article
- Export citation
Shigellosis outbreak among persons experiencing homelessness—San Diego County, California, October–December 2021
-
- Journal:
- Epidemiology & Infection / Volume 152 / 2024
- Published online by Cambridge University Press:
- 23 October 2023, e61
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Prevalence of Neisseria gonorrhoeae and Chlamydia trachomatis infections among adolescent men who have sex with men and transgender women in Salvador, Northeast Brazil
-
- Journal:
- Epidemiology & Infection / Volume 151 / 2023
- Published online by Cambridge University Press:
- 23 October 2023, e196
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The unified extropy and its versions in classical and Dempster–Shafer theories
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 23 October 2023, pp. 685-696
- Print publication:
- June 2024
-
- Article
- Export citation
On a retrial queue with negative customers, passive breakdown, and delayed repairs
-
- Journal:
- Probability in the Engineering and Informational Sciences / Volume 38 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 20 October 2023, pp. 428-447
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Spanning subdivisions in Dirac graphs
- Part of
-
- Journal:
- Combinatorics, Probability and Computing / Volume 33 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 20 October 2023, pp. 121-128
-
- Article
- Export citation
-
We show that for every
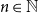 $n\in \mathbb N$ and
$n\in \mathbb N$ and 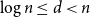 $\log n\le d\lt n$, if a graph
$\log n\le d\lt n$, if a graph 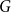 $G$ has
$G$ has 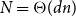 $N=\Theta (dn)$ vertices and minimum degree
$N=\Theta (dn)$ vertices and minimum degree 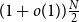 $(1+o(1))\frac{N}{2}$, then it contains a spanning subdivision of every
$(1+o(1))\frac{N}{2}$, then it contains a spanning subdivision of every  $n$-vertex
$n$-vertex 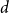 $d$-regular graph.
$d$-regular graph.
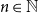
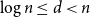
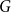
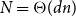
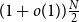

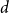
Construction of rating systems using global sensitivity analysis: A numerical investigation
-
- Journal:
- ASTIN Bulletin: The Journal of the IAA / Volume 54 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 19 October 2023, pp. 25-45
- Print publication:
- January 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
On Jensen-
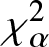 $\chi_{\alpha}^{2}$ divergence measure
$\chi_{\alpha}^{2}$ divergence measure
-
- Journal:
- Probability in the Engineering and Informational Sciences / Volume 38 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 19 October 2023, pp. 403-427
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
The purpose of this paper is twofold. The first part is to introduce relative-
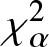 $\chi_{\alpha}^{2}$, Jensen-
$\chi_{\alpha}^{2}$, Jensen-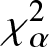 $\chi_{\alpha}^{2}$ and (p, w)-Jensen-
$\chi_{\alpha}^{2}$ and (p, w)-Jensen-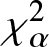 $\chi_{\alpha}^2$ divergence measures and then examine their properties. In addition, we also explore possible connections between these divergence measures and Jensen–Shannon entropy measure. In the second part, we introduce
$\chi_{\alpha}^2$ divergence measures and then examine their properties. In addition, we also explore possible connections between these divergence measures and Jensen–Shannon entropy measure. In the second part, we introduce 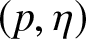 $(p,\eta)$-mixture model and then show it to be an optimal solution to three different optimization problems based on
$(p,\eta)$-mixture model and then show it to be an optimal solution to three different optimization problems based on 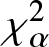 $\chi_{\alpha}^{2}$ divergence measure. We further study the relative-
$\chi_{\alpha}^{2}$ divergence measure. We further study the relative-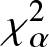 $\chi_{\alpha}^{2}$ divergence measure for escort and arithmetic mixture densities. We also provide some results associated with relative-
$\chi_{\alpha}^{2}$ divergence measure for escort and arithmetic mixture densities. We also provide some results associated with relative-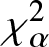 $\chi_{\alpha}^{2}$ divergence measure of mixed reliability systems. Finally, to demonstrate the usefulness of the Jensen-
$\chi_{\alpha}^{2}$ divergence measure of mixed reliability systems. Finally, to demonstrate the usefulness of the Jensen-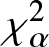 $\chi_{\alpha}^{2}$ divergence measure, we apply it to a real example in image processing and present some numerical results. Our findings in this regard show that the Jensen-
$\chi_{\alpha}^{2}$ divergence measure, we apply it to a real example in image processing and present some numerical results. Our findings in this regard show that the Jensen-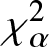 $\chi_{\alpha}^{2}$ is an effective criteria for quantifying the similarity between two images.
$\chi_{\alpha}^{2}$ is an effective criteria for quantifying the similarity between two images.
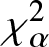
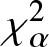
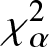
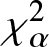
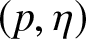
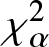
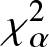
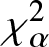
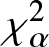
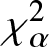
Prevalence and distribution of non-typhoidal Salmonella enterica serogroups and serovars isolated from normally sterile sites: A global systematic review
-
- Journal:
- Epidemiology & Infection / Volume 152 / 2024
- Published online by Cambridge University Press:
- 18 October 2023, e4
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Optimal VIX-linked structure for the target benefit pension plan
-
- Journal:
- ASTIN Bulletin: The Journal of the IAA / Volume 54 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 18 October 2023, pp. 75-93
- Print publication:
- January 2024
-
- Article
- Export citation
Risk sharing in equity-linked insurance products: Stackelberg equilibrium between an insurer and a reinsurer
-
- Journal:
- ASTIN Bulletin: The Journal of the IAA / Volume 54 / Issue 1 / January 2024
- Published online by Cambridge University Press:
- 18 October 2023, pp. 129-158
- Print publication:
- January 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
