Refine listing
Actions for selected content:
1416829 results in Open Access
The End of Tutela Mulierum
-
- Journal:
- The Journal of Roman Studies / Volume 114 / November 2024
- Published online by Cambridge University Press:
- 14 November 2024, pp. 85-103
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
IPO volume 54 issue 3 Cover and Front matter
-
- Journal:
- Italian Political Science Review / Rivista Italiana di Scienza Politica / Volume 54 / Issue 3 / November 2024
- Published online by Cambridge University Press:
- 14 January 2025, pp. f1-f2
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
Slavery on the Northern Frontier: A Stylus Tablet from Vindolanda
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
1 Introduction and overview
-
- Journal:
- Archaeological Reports / Volume 70 / November 2024
- Published online by Cambridge University Press:
- 30 December 2024, pp. 1-10
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Ottomans in the Caucasian Highlands: Recruitment of the Circassians and the Ottoman Mission in Anapa, 1812–1828
-
- Journal:
- International Journal of Middle East Studies / Volume 56 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 13 January 2025, pp. 645-662
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
A ‘Turk’ in eighteenth-century Dublin: the rise and fall of Doctor Achmet Borumborad
-
- Journal:
- Irish Historical Studies / Volume 48 / Issue 174 / November 2024
- Published online by Cambridge University Press:
- 13 May 2025, pp. 250-268
- Print publication:
- November 2024
-
- Article
- Export citation
LIS volume 55 Cover and Front matter
-
- Journal:
- Libyan Studies / Volume 55 / November 2024
- Published online by Cambridge University Press:
- 19 March 2025, pp. f1-f5
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
THE MENTAL ELEMENT OF ACCESSORY LIABILITY IN TORT
-
- Journal:
- The Cambridge Law Journal / Volume 83 / Issue 3 / November 2024
- Published online by Cambridge University Press:
- 07 January 2025, pp. 409-412
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Women, gender and the ancient economy: towards a feminist economic history of the ancient Greek world
-
- Journal:
- The Journal of Hellenic Studies / Volume 144 / November 2024
- Published online by Cambridge University Press:
- 18 November 2024, pp. 1-28
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Tachikawa's second conjecture, derived recollements, and gendo-symmetric algebras
- Part of
-
- Journal:
- Compositio Mathematica / Volume 160 / Issue 11 / November 2024
- Published online by Cambridge University Press:
- 27 December 2024, pp. 2704-2737
- Print publication:
- November 2024
-
- Article
- Export citation
-
Tachikawa's second conjecture for symmetric algebras is shown to be equivalent to indecomposable symmetric algebras not having any nontrivial stratifying ideals. The conjecture is also shown to be equivalent to the supremum of stratified ratios being less than
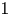 $1$, when taken over all indecomposable symmetric algebras. An explicit construction provides a series of counterexamples to Tachikawa's second conjecture from each (potentially existing) gendo-symmetric algebra that is a counterexample to Nakayama's conjecture. The results are based on establishing recollements of derived categories and on constructing new series of algebras.
$1$, when taken over all indecomposable symmetric algebras. An explicit construction provides a series of counterexamples to Tachikawa's second conjecture from each (potentially existing) gendo-symmetric algebra that is a counterexample to Nakayama's conjecture. The results are based on establishing recollements of derived categories and on constructing new series of algebras.
LSY volume 53 issue 5 Cover and Back matter
-
- Journal:
- Language in Society / Volume 53 / Issue 5 / November 2024
- Published online by Cambridge University Press:
- 20 December 2024, pp. b1-b5
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
Hybrid yoghurt: enhancing consumer acceptance by combining dairy and plant-based derivatives
-
- Journal:
- Journal of Dairy Research / Volume 91 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 04 March 2025, pp. 498-504
- Print publication:
- November 2024
-
- Article
- Export citation
The Interaction between International and Domestic Law, Aqua Nullius, and Water-mediated Claims in Canada
-
- Journal:
- Canadian Yearbook of International Law / Annuaire canadien de droit international / Volume 61 / November 2024
- Published online by Cambridge University Press:
- 12 December 2024, pp. 206-236
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
‘I can’t segregate myself’: self-narrating and ‘small boundary’ work in Nairobi’s ghettos
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Sire association with intramammary infection and clinical mastitis in a Holstein-Friesian × Jersey crossbred pedigree herd
-
- Journal:
- Journal of Dairy Research / Volume 91 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 19 May 2025, pp. 410-419
- Print publication:
- November 2024
-
- Article
- Export citation
International Book Essay Section
-
- Journal:
- Law & Social Inquiry / Volume 49 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 04 December 2024, p. 2581
- Print publication:
- November 2024
-
- Article
- Export citation
The great convergence: post-Cold War transitions to hybrid regimes across waves and ebbs
-
- Journal:
- Italian Political Science Review / Rivista Italiana di Scienza Politica / Volume 54 / Issue 3 / November 2024
- Published online by Cambridge University Press:
- 05 December 2024, pp. 248-256
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
SJT volume 77 issue 4 Cover and Back matter
-
- Journal:
- Scottish Journal of Theology / Volume 77 / Issue 4 / November 2024
- Published online by Cambridge University Press:
- 27 December 2024, pp. b1-b2
- Print publication:
- November 2024
-
- Article
-
- You have access
- Export citation
A new lichen and lichenicolous fungus from Larix laricina in patterned fens of boreal North America
-
- Journal:
- The Lichenologist / Volume 56 / Issue 6 / November 2024
- Published online by Cambridge University Press:
- 31 December 2024, pp. 379-392
- Print publication:
- November 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
