Refine search
Actions for selected content:
115 results
On the dynamics of a diffusive SEIRS epidemic model
- Part of
-
- Journal:
- European Journal of Applied Mathematics , First View
- Published online by Cambridge University Press:
- 13 April 2026, pp. 1-45
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
This work investigates the dynamics of positive classical solutions to a diffusive susceptible-exposed-infected-recovered-susceptible epidemic model with a mass-action incidence mechanism in spatially heterogeneous environments. Under minimal assumptions on the initial data, the global existence of classical solutions is established. Moreover, the eventual boundedness of these solutions is proved when either the spatial domain has dimension five or lower or the susceptible and exposed subpopulations share the same diffusion rate. Next, we define the basic reproduction number,
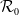 $\mathcal{R}_0$, and demonstrate that the disease-free equilibrium is globally stable when
$\mathcal{R}_0$, and demonstrate that the disease-free equilibrium is globally stable when 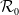 $\mathcal{R}_0$ is sufficiently small. However, due to the complex interaction between population movement and spatial variation in transmission rates, we find that the disease may persist even when
$\mathcal{R}_0$ is sufficiently small. However, due to the complex interaction between population movement and spatial variation in transmission rates, we find that the disease may persist even when 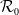 $\mathcal{R}_0$ is slightly less than one. In such cases, we show that the system admits at least two endemic equilibrium (EE) solutions, an outcome not observed under the frequency-dependent incidence mechanism. These results highlight the significant influence of the transmission mechanism on disease dynamics. Furthermore, we examine the spatial profiles of the EE solutions when diffusion rates are small. Our analysis suggests that limiting the movement of the susceptible population can significantly reduce disease prevalence, provided that the total population remains below a specific threshold. In contrast, restricting the movement of the infected, exposed, or recovered populations alone may not eradicate the disease. Overall, our findings provide important insights into the spatial dynamics of infectious diseases and may offer guidance for developing and implementing effective containment strategies.
$\mathcal{R}_0$ is slightly less than one. In such cases, we show that the system admits at least two endemic equilibrium (EE) solutions, an outcome not observed under the frequency-dependent incidence mechanism. These results highlight the significant influence of the transmission mechanism on disease dynamics. Furthermore, we examine the spatial profiles of the EE solutions when diffusion rates are small. Our analysis suggests that limiting the movement of the susceptible population can significantly reduce disease prevalence, provided that the total population remains below a specific threshold. In contrast, restricting the movement of the infected, exposed, or recovered populations alone may not eradicate the disease. Overall, our findings provide important insights into the spatial dynamics of infectious diseases and may offer guidance for developing and implementing effective containment strategies.
The final outcome of SIR epidemics with infectivity profiles
- Part of
-
- Journal:
- Journal of Applied Probability , First View
- Published online by Cambridge University Press:
- 31 March 2026, pp. 1-23
-
- Article
- Export citation
-
A stochastic model for the spread of an SIR (susceptible
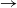 $\to$ infective
$\to$ infective 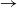 $\to$ removed) epidemic is considered. Infectives have independent and identically distributed infectivity profiles, which describe their infectiousness as a function of time since infection. The individual-to-individual infection rate depends also on the number of susceptibles present in the population. Exact results are derived for the distribution of statistics defined on the final outcome of the epidemic, including its final size. These are proved by using a generalisation of a Sellke construction to show that the distribution of the final outcome of the epidemic is the same as that of an associated discrete-time epidemic process, in which infectives are considered one at a time, and exploiting connection with death processes to analyse the final outcome of the latter. The results generalise easily to multipopulation epidemics.
$\to$ removed) epidemic is considered. Infectives have independent and identically distributed infectivity profiles, which describe their infectiousness as a function of time since infection. The individual-to-individual infection rate depends also on the number of susceptibles present in the population. Exact results are derived for the distribution of statistics defined on the final outcome of the epidemic, including its final size. These are proved by using a generalisation of a Sellke construction to show that the distribution of the final outcome of the epidemic is the same as that of an associated discrete-time epidemic process, in which infectives are considered one at a time, and exploiting connection with death processes to analyse the final outcome of the latter. The results generalise easily to multipopulation epidemics.
Infinite dimensional metapopulation SIS model with generalized incidence rate
- Part of
-
- Journal:
- Canadian Journal of Mathematics , First View
- Published online by Cambridge University Press:
- 29 January 2026, pp. 1-43
-
- Article
- Export citation
Global dynamics of a degenerate reaction-diffusion cholera model with phage-bacteria interaction in a heterogeneous environment
- Part of
-
- Journal:
- European Journal of Applied Mathematics , First View
- Published online by Cambridge University Press:
- 22 September 2025, pp. 1-32
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
To investigate multiple effects of the interaction between V. cholerae and phage on cholera transmission, we propose a degenerate reaction-diffusion model with different dispersal rates, which incorporates a short-lived hyperinfectious (HI vibrios) state of V. cholerae and lower-infectious (LI vibrios) state of V. cholerae. Our main purpose is to investigate the existence and stability analysis of multi-class boundary steady states, which is much more complicated and challenging than the case when the boundary steady state is unique. In a spatially heterogeneous case, the basic reproduction number
 $\mathscr{R}_{0}$ is defined as the spectral radius of the sum of two linear operators associated with HI vibrios infection and LI vibrios infection. If
$\mathscr{R}_{0}$ is defined as the spectral radius of the sum of two linear operators associated with HI vibrios infection and LI vibrios infection. If  $\mathscr{R}_{0}\leq 1$, the disease-free steady state is globally asymptotically stable. If
$\mathscr{R}_{0}\leq 1$, the disease-free steady state is globally asymptotically stable. If  $\mathscr{R}_{0}\gt 1$, the uniform persistence of phage-free model, as well as the existence of the phage-free steady state, are established. In a spatially homogeneous case, when
$\mathscr{R}_{0}\gt 1$, the uniform persistence of phage-free model, as well as the existence of the phage-free steady state, are established. In a spatially homogeneous case, when  $\ \;\widetilde{\!\!\!\mathscr{R}}_{0}\gt 1$, the global asymptotic stability of phage-free steady state and the uniform persistence of the phage-present model are discussed under some additional conditions. The mathematical approach here has wide applications in degenerate Partial Differential Equations.
$\ \;\widetilde{\!\!\!\mathscr{R}}_{0}\gt 1$, the global asymptotic stability of phage-free steady state and the uniform persistence of the phage-present model are discussed under some additional conditions. The mathematical approach here has wide applications in degenerate Partial Differential Equations.




Periodic dynamics of a general switching dynamical system
- Part of
-
- Journal:
- European Journal of Applied Mathematics , First View
- Published online by Cambridge University Press:
- 08 August 2025, pp. 1-15
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Epidemic models with digital and manual contact tracing
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 57 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 09 June 2025, pp. 1430-1455
- Print publication:
- December 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
We analyse a Markovian SIR epidemic model where individuals either recover naturally or are diagnosed, leading to isolation and potential contact tracing. Our focus is on digital contact tracing via a tracing app, considering both its standalone use and its combination with manual tracing. We prove that as the population size n grows large, the epidemic process converges to a limiting process, which, unlike with typical epidemic models, is not a branching process due to dependencies created by contact tracing. However, by grouping to-be-traced individuals into macro-individuals, we derive a multi-type branching process interpretation, allowing computation of the reproduction number R. This is then converted to an individual reproduction number
 $R^\mathrm{(ind)}$, which, in contrast to R, decays monotonically with the fraction of app-users, while both share the same threshold at 1. Finally, we compare digital (only) contact tracing and manual (only) contact tracing, proving that the critical fraction of app-users,
$R^\mathrm{(ind)}$, which, in contrast to R, decays monotonically with the fraction of app-users, while both share the same threshold at 1. Finally, we compare digital (only) contact tracing and manual (only) contact tracing, proving that the critical fraction of app-users,  $\pi_{\mathrm{c}}$, required for
$\pi_{\mathrm{c}}$, required for  $R=1$ is higher than the critical fraction manually contact-traced,
$R=1$ is higher than the critical fraction manually contact-traced,  $p_{\mathrm{c}}$, for manual tracing.
$p_{\mathrm{c}}$, for manual tracing.




Cutoff for the logistic SIS epidemic model with self-infection
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 57 / Issue 4 / December 2025
- Published online by Cambridge University Press:
- 04 June 2025, pp. 1456-1483
- Print publication:
- December 2025
-
- Article
- Export citation
Optimal control of a reaction-diffusion epidemic model with non-compliance
- Part of
-
- Journal:
- European Journal of Applied Mathematics / Volume 37 / Issue 2 / April 2026
- Published online by Cambridge University Press:
- 14 April 2025, pp. 313-338
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The real-time growth rate of stochastic epidemics on random intersection graphs
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 62 / Issue 3 / September 2025
- Published online by Cambridge University Press:
- 24 March 2025, pp. 1132-1155
- Print publication:
- September 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The number of individuals alive in a branching process given only times of deaths
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 57 / Issue 3 / September 2025
- Published online by Cambridge University Press:
- 31 January 2025, pp. 871-906
- Print publication:
- September 2025
-
- Article
- Export citation
Propagation dynamics for an epidemic patch model with variable incubation period
- Part of
-
- Journal:
- European Journal of Applied Mathematics / Volume 36 / Issue 5 / October 2025
- Published online by Cambridge University Press:
- 18 December 2024, pp. 948-971
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Travelling wave solutions and spreading speeds for a scalar age-structured equation with nonlocal diffusion
- Part of
-
- Journal:
- European Journal of Applied Mathematics / Volume 36 / Issue 4 / August 2025
- Published online by Cambridge University Press:
- 15 November 2024, pp. 856-878
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
On degenerate reaction-diffusion epidemic models with mass action or standard incidence mechanism
- Part of
-
- Journal:
- European Journal of Applied Mathematics / Volume 35 / Issue 5 / October 2024
- Published online by Cambridge University Press:
- 22 January 2024, pp. 634-661
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Recurrent and chaotic outbreaks in SIR model
-
- Journal:
- European Journal of Applied Mathematics / Volume 35 / Issue 5 / October 2024
- Published online by Cambridge University Press:
- 18 January 2024, pp. 662-674
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
A queueing system with an SIR-type infection
- Part of
-
- Journal:
- Probability in the Engineering and Informational Sciences / Volume 38 / Issue 3 / July 2024
- Published online by Cambridge University Press:
- 16 January 2024, pp. 559-578
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
SIR model with social gatherings
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 61 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 15 January 2024, pp. 667-684
- Print publication:
- June 2024
-
- Article
- Export citation
Strong convergence of an epidemic model with mixing groups
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 56 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 01 September 2023, pp. 430-463
- Print publication:
- June 2024
-
- Article
- Export citation
-
We consider an SIR (susceptible
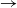 $\to$ infective
$\to$ infective 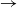 $\to$ recovered) epidemic in a closed population of size n, in which infection spreads via mixing events, comprising individuals chosen uniformly at random from the population, which occur at the points of a Poisson process. This contrasts sharply with most epidemic models, in which infection is spread purely by pairwise interaction. A sequence of epidemic processes, indexed by n, and an approximating branching process are constructed on a common probability space via embedded random walks. We show that under suitable conditions the process of infectives in the epidemic process converges almost surely to the branching process. This leads to a threshold theorem for the epidemic process, where a major outbreak is defined as one that infects at least
$\to$ recovered) epidemic in a closed population of size n, in which infection spreads via mixing events, comprising individuals chosen uniformly at random from the population, which occur at the points of a Poisson process. This contrasts sharply with most epidemic models, in which infection is spread purely by pairwise interaction. A sequence of epidemic processes, indexed by n, and an approximating branching process are constructed on a common probability space via embedded random walks. We show that under suitable conditions the process of infectives in the epidemic process converges almost surely to the branching process. This leads to a threshold theorem for the epidemic process, where a major outbreak is defined as one that infects at least 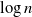 $\log n$ individuals. We show further that there exists
$\log n$ individuals. We show further that there exists 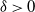 $\delta \gt 0$, depending on the model parameters, such that the probability that a major outbreak has size at least
$\delta \gt 0$, depending on the model parameters, such that the probability that a major outbreak has size at least 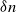 $\delta n$ tends to one as
$\delta n$ tends to one as 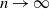 $n \to \infty$.
$n \to \infty$.
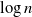
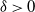
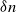
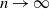
Branching processes in random environments with thresholds
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 56 / Issue 2 / June 2024
- Published online by Cambridge University Press:
- 22 August 2023, pp. 495-544
- Print publication:
- June 2024
-
- Article
- Export citation
-
Motivated by applications to COVID dynamics, we describe a model of a branching process in a random environment
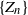 $\{Z_n\}$ whose characteristics change when crossing upper and lower thresholds. This introduces a cyclical path behavior involving periods of increase and decrease leading to supercritical and subcritical regimes. Even though the process is not Markov, we identify subsequences at random time points
$\{Z_n\}$ whose characteristics change when crossing upper and lower thresholds. This introduces a cyclical path behavior involving periods of increase and decrease leading to supercritical and subcritical regimes. Even though the process is not Markov, we identify subsequences at random time points 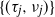 $\{(\tau_j, \nu_j)\}$—specifically the values of the process at crossing times, viz.
$\{(\tau_j, \nu_j)\}$—specifically the values of the process at crossing times, viz. 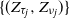 $\{(Z_{\tau_j}, Z_{\nu_j})\}$—along which the process retains the Markov structure. Under mild moment and regularity conditions, we establish that the subsequences possess a regenerative structure and prove that the limiting normal distributions of the growth rates of the process in supercritical and subcritical regimes decouple. For this reason, we establish limit theorems concerning the length of supercritical and subcritical regimes and the proportion of time the process spends in these regimes. As a byproduct of our analysis, we explicitly identify the limiting variances in terms of the functionals of the offspring distribution, threshold distribution, and environmental sequences.
$\{(Z_{\tau_j}, Z_{\nu_j})\}$—along which the process retains the Markov structure. Under mild moment and regularity conditions, we establish that the subsequences possess a regenerative structure and prove that the limiting normal distributions of the growth rates of the process in supercritical and subcritical regimes decouple. For this reason, we establish limit theorems concerning the length of supercritical and subcritical regimes and the proportion of time the process spends in these regimes. As a byproduct of our analysis, we explicitly identify the limiting variances in terms of the functionals of the offspring distribution, threshold distribution, and environmental sequences.
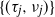
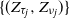
Migration–contagion processes
- Part of
-
- Journal:
- Advances in Applied Probability / Volume 56 / Issue 1 / March 2024
- Published online by Cambridge University Press:
- 18 August 2023, pp. 71-105
- Print publication:
- March 2024
-
- Article
- Export citation
-
Consider the following migration process based on a closed network of N queues with
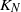 $K_N$ customers. Each station is a
$K_N$ customers. Each station is a  $\cdot$/M/
$\cdot$/M/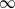 $\infty$ queue with service (or migration) rate
$\infty$ queue with service (or migration) rate 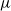 $\mu$. Upon departure, a customer is routed independently and uniformly at random to another station. In addition to migration, these customers are subject to a susceptible–infected–susceptible (SIS) dynamics. That is, customers are in one of two states: I for infected, or S for susceptible. Customers can swap their state either from I to S or from S to I only in stations. More precisely, at any station, each susceptible customer becomes infected with the instantaneous rate
$\mu$. Upon departure, a customer is routed independently and uniformly at random to another station. In addition to migration, these customers are subject to a susceptible–infected–susceptible (SIS) dynamics. That is, customers are in one of two states: I for infected, or S for susceptible. Customers can swap their state either from I to S or from S to I only in stations. More precisely, at any station, each susceptible customer becomes infected with the instantaneous rate 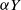 $\alpha Y$ if there are Y infected customers in the station, whereas each infected customer recovers and becomes susceptible with rate
$\alpha Y$ if there are Y infected customers in the station, whereas each infected customer recovers and becomes susceptible with rate 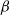 $\beta$. We let N tend to infinity and assume that
$\beta$. We let N tend to infinity and assume that 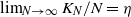 $\lim_{N\to \infty} K_N/N= \eta $, where
$\lim_{N\to \infty} K_N/N= \eta $, where 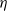 $\eta$ is a positive constant representing the customer density. The main problem of interest concerns the set of parameters of such a system for which there exists a stationary regime where the epidemic survives in the limiting system. The latter limit will be referred to as the thermodynamic limit. We use coupling and stochastic monotonicity arguments to establish key properties of the associated Markov processes, which in turn allow us to give the structure of the phase transition diagram of this thermodynamic limit with respect to
$\eta$ is a positive constant representing the customer density. The main problem of interest concerns the set of parameters of such a system for which there exists a stationary regime where the epidemic survives in the limiting system. The latter limit will be referred to as the thermodynamic limit. We use coupling and stochastic monotonicity arguments to establish key properties of the associated Markov processes, which in turn allow us to give the structure of the phase transition diagram of this thermodynamic limit with respect to 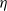 $\eta$. The analysis of the Kolmogorov equations of this SIS model reduces to that of a wave-type PDE for which we have found no explicit solution. This plain SIS model is one among several companion stochastic processes that exhibit both random migration and contagion. Two of them are discussed in the present paper as they provide variants to the plain SIS model as well as some bounds and approximations. These two variants are the departure-on-change-of-state (DOCS) model and the averaged-infection-rate (AIR) model, which both admit closed-form solutions. The AIR system is a classical mean-field model where the infection mechanism based on the actual population of infected customers is replaced by a mechanism based on some empirical average of the number of infected customers in all stations. The latter admits a product-form solution. DOCS features accelerated migration in that each change of SIS state implies an immediate departure. This model leads to another wave-type PDE that admits a closed-form solution. In this text, the main focus is on the closed stochastic networks and their limits. The open systems consisting of a single station with Poisson input are instrumental in the analysis of the thermodynamic limits and are also of independent interest. This class of SIS dynamics has incarnations in virtually all queueing networks of the literature.
$\eta$. The analysis of the Kolmogorov equations of this SIS model reduces to that of a wave-type PDE for which we have found no explicit solution. This plain SIS model is one among several companion stochastic processes that exhibit both random migration and contagion. Two of them are discussed in the present paper as they provide variants to the plain SIS model as well as some bounds and approximations. These two variants are the departure-on-change-of-state (DOCS) model and the averaged-infection-rate (AIR) model, which both admit closed-form solutions. The AIR system is a classical mean-field model where the infection mechanism based on the actual population of infected customers is replaced by a mechanism based on some empirical average of the number of infected customers in all stations. The latter admits a product-form solution. DOCS features accelerated migration in that each change of SIS state implies an immediate departure. This model leads to another wave-type PDE that admits a closed-form solution. In this text, the main focus is on the closed stochastic networks and their limits. The open systems consisting of a single station with Poisson input are instrumental in the analysis of the thermodynamic limits and are also of independent interest. This class of SIS dynamics has incarnations in virtually all queueing networks of the literature.
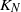

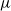
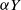
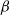
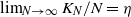
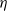
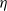
SIR epidemics driven by Feller processes
- Part of
-
- Journal:
- Journal of Applied Probability / Volume 60 / Issue 4 / December 2023
- Published online by Cambridge University Press:
- 02 May 2023, pp. 1293-1316
- Print publication:
- December 2023
-
- Article
- Export citation
-
We consider a stochastic SIR (susceptible
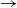 $\rightarrow$ infective
$\rightarrow$ infective 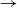 $\rightarrow$ removed) model in which the infectious periods are modulated by a collection of independent and identically distributed Feller processes. Each infected individual is associated with one of these processes, the trajectories of which determine the duration of his infectious period, his contamination rate, and his type of removal (e.g. death or immunization). We use a martingale approach to derive the distribution of the final epidemic size and severity for this model and provide some general examples. Next, we focus on a single infected individual facing a given number of susceptibles, and we determine the distribution of his outcome (number of contaminations, severity, type of removal). Using a discrete-time formulation of the model, we show that this distribution also provides us with an alternative, more stable method to compute the final epidemic outcome distribution.
$\rightarrow$ removed) model in which the infectious periods are modulated by a collection of independent and identically distributed Feller processes. Each infected individual is associated with one of these processes, the trajectories of which determine the duration of his infectious period, his contamination rate, and his type of removal (e.g. death or immunization). We use a martingale approach to derive the distribution of the final epidemic size and severity for this model and provide some general examples. Next, we focus on a single infected individual facing a given number of susceptibles, and we determine the distribution of his outcome (number of contaminations, severity, type of removal). Using a discrete-time formulation of the model, we show that this distribution also provides us with an alternative, more stable method to compute the final epidemic outcome distribution.