Refine listing
Actions for selected content:
1418543 results in Open Access
The Liar’s Dividend: Can Politicians Claim Misinformation to Evade Accountability?
-
- Journal:
- American Political Science Review / Volume 119 / Issue 1 / February 2025
- Published online by Cambridge University Press:
- 20 February 2024, pp. 71-90
- Print publication:
- February 2025
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Being Open-Minded about Open-Mindedness
-
- Journal:
- Philosophy / Volume 99 / Issue 2 / April 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 191-221
- Print publication:
- April 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Parameterized complexity of weighted team definability
-
- Journal:
- Mathematical Structures in Computer Science / Volume 34 / Issue 5 / May 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 375-389
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
In this article, we study the complexity of weighted team definability for logics with team semantics. This problem is a natural analog of one of the most studied problems in parameterized complexity, the notion of weighted Fagin-definability, which is formulated in terms of satisfaction of first-order formulas with free relation variables. We focus on the parameterized complexity of weighted team definability for a fixed formula
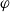 $\varphi$ of central team-based logics. Given a first-order structure
$\varphi$ of central team-based logics. Given a first-order structure 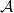 $\mathcal{A}$ and the parameter value
$\mathcal{A}$ and the parameter value 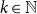 $k\in \mathbb N$ as input, the question is to determine whether
$k\in \mathbb N$ as input, the question is to determine whether 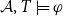 $\mathcal{A},T\models \varphi$ for some team T of size k. We show several results on the complexity of this problem for dependence, independence, and inclusion logic formulas. Moreover, we also relate the complexity of weighted team definability to the complexity classes in the well-known W-hierarchy as well as paraNP.
$\mathcal{A},T\models \varphi$ for some team T of size k. We show several results on the complexity of this problem for dependence, independence, and inclusion logic formulas. Moreover, we also relate the complexity of weighted team definability to the complexity classes in the well-known W-hierarchy as well as paraNP.
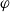
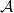
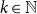
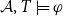
Effect of vegetable consumption on risk of gastric cancer: a systematic review and multi-level meta-analysis of prospective studies
-
- Journal:
- Nutrition Research Reviews / Volume 38 / Issue 1 / June 2025
- Published online by Cambridge University Press:
- 20 February 2024, pp. 192-201
-
- Article
- Export citation
Translation of the Preference-Based Amyotrophic Lateral Sclerosis Scale into French
-
- Journal:
- Canadian Journal of Neurological Sciences / Volume 52 / Issue 1 / January 2025
- Published online by Cambridge University Press:
- 20 February 2024, pp. 129-131
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
PHO volume 39 issue 4 Cover and Front matter
-
- Article
-
- You have access
- Export citation
MRF volume 15 issue 10 Cover and Back matter
-
- Journal:
- International Journal of Microwave and Wireless Technologies / Volume 15 / Issue 10 / December 2023
- Published online by Cambridge University Press:
- 20 February 2024, p. b1
-
- Article
-
- You have access
- Export citation
Oscillation-free point-to-point motions of planar differentially flat under-actuated robots: a Laplace transform method
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Differentially flat under-actuated robots are characterized by more degrees of freedom (DOF) than actuators: this makes possible the design of lightweight cheap robots with high dexterity. The main issue of such robots is the control of the passive joint, which requires accurate dynamic modeling of the robot.
Friction is usually discarded to simplify the models, especially in the case of low-speed trajectories. However, this simplification leads to oscillations of the end-effector about the final position, which are incompatible with fast and accurate motions.
This paper focuses on planar
 $n$-DOF serial robotic arms with
$n$-DOF serial robotic arms with 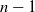 $n-1$ actuated rotational joints plus one final passive rotational joint with stiffness and friction properties. These robots, if properly balanced, are differentially flat. When the non-actuated joint can be considered frictionless, differentially flat robots can be controlled in open loop, calculating the motor torques demanded by point-to-point motions. This paper extends the open-loop control to robots with a passive joint with viscous friction adopting a Laplace transform method. This method can be adopted by exploiting the particular structure of the equations of motion of differentially flat under-actuated robots in which the last equations are linear. Analytical expressions of the motor torques are obtained. The work is enriched by an experimental validation of a
$n-1$ actuated rotational joints plus one final passive rotational joint with stiffness and friction properties. These robots, if properly balanced, are differentially flat. When the non-actuated joint can be considered frictionless, differentially flat robots can be controlled in open loop, calculating the motor torques demanded by point-to-point motions. This paper extends the open-loop control to robots with a passive joint with viscous friction adopting a Laplace transform method. This method can be adopted by exploiting the particular structure of the equations of motion of differentially flat under-actuated robots in which the last equations are linear. Analytical expressions of the motor torques are obtained. The work is enriched by an experimental validation of a 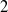 $2$-DOF under-actuated robot and by numerical simulations of the
$2$-DOF under-actuated robot and by numerical simulations of the 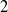 $2$- and
$2$- and 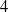 $4$-DOF robots showing the suppression of unwanted oscillations.
$4$-DOF robots showing the suppression of unwanted oscillations.

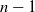
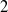
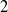
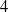
Catheter-associated urinary tract infections (CAUTIs) and non-CAUTI hospital-onset urinary tract infections: Relative burden, cost, outcomes and related hospital-onset bacteremia and fungemia infections
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 45 / Issue 7 / July 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 864-871
- Print publication:
- July 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The onset of mental health disparities in sexual minority and majority youth: evidence from the UK Millennium Cohort Study
-
- Journal:
- Development and Psychopathology / Volume 37 / Issue 1 / February 2025
- Published online by Cambridge University Press:
- 20 February 2024, pp. 504-514
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Design of miniaturized single and dual-band bandpass filters using diamond-shaped coupled line resonator for next-generation wireless systems – ERRATUM
-
- Journal:
- International Journal of Microwave and Wireless Technologies / Volume 15 / Issue 10 / December 2023
- Published online by Cambridge University Press:
- 20 February 2024, p. 1809
-
- Article
-
- You have access
- HTML
- Export citation
Interplay between polygenic variants related immune response and lifestyle factors mitigate the chances of stroke in a genome-wide association study
-
- Journal:
- British Journal of Nutrition / Volume 131 / Issue 10 / 28 May 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 1813-1826
- Print publication:
- 28 May 2024
-
- Article
-
- You have access
- HTML
- Export citation
Palmer-Rubin Brian. (2022). Evading the Patronage Trap. Interest Representation in Mexico. Ann Arbor: University of Michigan Press. Illustrations, bibliography, index, appendices, 323 pp.; hardcover US$90.00, paperback US$42.95, read online with open access
-
- Journal:
- Latin American Politics and Society / Volume 66 / Issue 3 / August 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 207-210
-
- Article
- Export citation
Dreaming A Better World for Animals: A Review of David Peña-Guzmán’s When Animals Dream: The Hidden World of Animal Consciousness, 2022, 259 pp. ISBN 9780691220093.
-
- Journal:
- Cambridge Quarterly of Healthcare Ethics / Volume 33 / Issue 4 / October 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 598-600
-
- Article
- Export citation
HISTORY OF ECONOMIC THOUGHT’S PLACE IN MACROECONOMICS REVISITED
-
- Journal:
- Journal of the History of Economic Thought / Volume 46 / Issue 4 / December 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 509-517
- Print publication:
- December 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Susceptibility of healthcare personnel with severe acute respiratory coronavirus virus 2 (SARS-CoV-2) hybrid immunity to XBB lineage reinfection
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 45 / Issue 6 / June 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 781-784
- Print publication:
- June 2024
-
- Article
- Export citation
‘When it goes well, it works fantastically’: motivations to train and their impact on the practice of CBT
-
- Journal:
- The Cognitive Behaviour Therapist / Volume 17 / 2024
- Published online by Cambridge University Press:
- 20 February 2024, e6
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The thermophilic sea anemone Telmatactis cricoides (Cnidaria, Hexacorallia) in the western Mediterranean: filling gaps in the knowledge of the distribution
-
- Journal:
- Journal of the Marine Biological Association of the United Kingdom / Volume 104 / 2024
- Published online by Cambridge University Press:
- 20 February 2024, e9
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Measures of maximal entropy of bounded density shifts
- Part of
-
- Journal:
- Ergodic Theory and Dynamical Systems / Volume 44 / Issue 10 / October 2024
- Published online by Cambridge University Press:
- 20 February 2024, pp. 2960-2974
- Print publication:
- October 2024
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Thomas M. Truxes. The Overseas Trade of British America: A Narrative History. New Haven: Yale University Press, 2021. Pp. 464. $40.00 (cloth).
-
- Journal:
- Journal of British Studies / Volume 62 / Issue 4 / October 2023
- Published online by Cambridge University Press:
- 19 February 2024, pp. 1077-1079
-
- Article
- Export citation
